Fig. 1.1
Clinical features of pemphigus vulgaris. Flaccid blisters, erosions and crusting on the face (a) and back (b) of a 55-year-old man
Pemphigus vegetans, a rare subtype of PV, is typified by a localized vegetative or papillomatous response. There are two types of pemphigus vegetans: the Hallopeau type and the Neumann type [3]. In pemphigus vegetans of Hallopeau, pustular lesions predominantly involve the folds and heal into localized verrucous, hyperkeratotic plaques. Pemphigus vegetans of Neumann is more extensive, characterized by periorificial papillomas and results in the formation of excess granulation tissue.
In 1964, Beutner and Jordan demonstrated the presence of anti-epidermal antibodies in the serum of pemphigus patients [4]. Anhalt et al. confirmed that passive transfer of PV immunoglobulin G (IgG) induced a pemphigus phenotype in neonatal mice [5]. Circulating IgG autoantibodies against the cadherins, desmogleins 1 (Dsg1) and 3 (Dsg3), were subsequently characterized [6]. By 1997, Koch et al. established that the disruption of desmoglein 3 in mice resulted in the loss of keratinocyte cell-cell adhesion and a PV phenotype [7]. The collective data support the pathogenicity of these antibodies in pemphigus patients.
PV is a complex polygenic disorder involving multiple genetic loci, many of which remain unknown. Association studies link HLA class II genes to PV, as over 95 % of patients carry either the DRB1*0402 or DQB1*0503 alleles [2]. Though rare, familial cases of the disease have been reported [8].
Up to 25 % of patients with pemphigus have another underlying immunologic disease [9]. Pemphigus is associated with autoimmune thyroid disease, type I diabetes, rheumatoid arthritis and systemic lupus erythematosus [10]. Further, the association between pemphigus and myasthenia gravis is well established [11]. These findings suggest that common genetic factors from clinically distinct disorders may underlie the susceptibility to autoimmune disease.
Genetic predisposition alone is not sufficient to cause the development of PV. Environmental factors seem to be required to initiate and perpetuate the disease process. However, an inducing agent cannot be identified in most patients. Occasionally, drugs, physical agents, contact allergens, viral infections, vaccinations, and diet have been implicated in the disease [11]. For instance, drug-induced pemphigus may occur with thiols (i.e., penicillamine, captopril), phenols (i.e., aspirin, rifampin) and non-thiol, non-phenol drugs (i.e., non-steroidal anti-inflammatories, nifedipine) [11].
If left untreated, PV has a mortality rate ranging from 60 to 90 % [12]. Overwhelming sepsis, fluid and electrolyte imbalances, impaired thermoregulation, as well as cardiac and renal failure are possible life-threatening complications of the disease. Systemic corticosteroids and adjuvant therapies have reduced the mortality rate of PV patients to approximately 10 %; yet, treatment-related complications are now the leading cause of morbidity and mortality. By understanding the molecular mechanisms that underlie pemphigus, researchers are developing novel targeted therapies for the management of PV.
Diagnosis
During the initial clinical encounter, the physician should look for signs and symptoms to support a diagnosis of pemphigus. A thorough evaluation for potential risk factors, triggers and comorbidities must be elicited. Validated scoring systems, such as the Autoimmune Bullous Skin Intensity and Severity Score (ABSIS) and the Pemphigus Disease Area Index (PDAI), may be used to measure the extent and distribution of lesions.
To confirm the diagnosis of PV, biopsies must be performed for both routine pathology and direct immunofluorescence (DIF). A 4.0 mm punch excision from an early, small vesicle or the periphery of a larger blister should be obtained for histopathologic analysis. Routine histology reveals loss of cellular cohesion (acantholysis) in the suprabasilar layer of the epithelium (Fig. 1.2a). A classical “tombstone” appearance of basal keratinocytes is commonly described (Fig. 1.2b). Direct immunofluorescence of perilesional skin demonstrates intercellular deposition of IgG and/or C3. Since antibodies correlate with disease activity in most patients, indirect immunofluorescence (IIF) assays are frequently used to semi-quantitatively measure circulating antibody levels. Using monkey or guinea pig esophagus as a substrate for IIF, an intercellular staining pattern may be visualized. The findings resemble a ‘chicken-wire’, ‘honeycomb’ or ‘fishnet’ appearance (Fig. 1.2c). Enzyme-linked immunosorbent assays (ELISA) are a more sensitive method for measuring antibodies to desmoglein 1 and desmoglein 3. Lastly, immunoblot and immunoprecipitation may be used to identify specific autoantibody profiles.
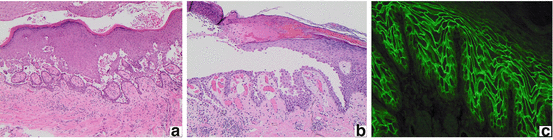
Fig. 1.2
Immunohistology of pemphigus vulgaris. Suprabasilar clefting, blister formation (a) and acantholysis (b) in a “tombstone” pattern (H&E, 100× magnification). Circulating intercellular IgG autoantibodies revealing a “chicken-wire,” “honey-comb,” or “fishnet” pattern on indirect immunofluorescence (c)
Management
A multidisciplinary approach is required to optimize patient care and outcomes. An experienced dermatologist must work closely with the patient’s general practitioner. Other specialists may play a supportive role for PV patients, including oral pathologists, otolaryngologists, ophthalmologists, gynecologists, urologists, internists and psychiatrists, among others [13].
Allied health care professionals often work with patients to minimize their co-morbidities. Educated wound care specialists are particularly helpful in providing non-adhesive dressing recommendations. Proper dental care and good oral hygiene are required. A dietician may provide nutritional support for patients with severe oral disease and resultant malnutrition, as well as those with steroid-related complications (i.e., diabetes, hypertension, obesity). If needed, analgesics should be ordered to ensure adequate pain control. Infections should be promptly recognized and treated aggressively.
If a potential triggering agent is identified on history, every effort must be made to eliminate the precipitating factor. As drug-induced pemphigus is a well-described phenomenon, a close review of the patient’s medication history is required. Other reported exogenous triggers include: (1) infections, such as herpes simplex virus; (2) physical agents, such as ultraviolet or ionizing radiation; (3) contact allergens, such as 1,3-dichloropropene; and (4) dietary factors, such as members of the Allium species (i.e., garlic, leeks, onions, chives) [2].
An extensive work-up is required prior to initiating corticosteroid and/or immunosuppressive therapies [13]. Recommended investigations include:
Complete blood count and differential;
Serum electrolytes, creatinine, urea;
Liver panel;
Fasting glucose, cholesterol and triglycerides;
Hepatitis B, Hepatitis C and HIV serologies;
Serum β−HCG on all woman of childbearing age;
Screening Mantoux test (or quantiFERON® test) for tuberculosis;
Chest X-ray;
Baseline bone density;
Baseline ocular examination.
If indicated, additional bloodwork may be requested to determine a patient’s candidacy for select therapies. For instance, thiopurine methyltransferase (TPMT) or glucose-6-phosphate dehydrogenase (G6PD) should be ordered prior to initiating treatment with azathioprine or dapsone, respectively. Further screening bloodwork and/or radiologic investigations should be considered on a case-by-case basis. Vaccinations (i.e., seasonal influenza, H1N1, tetanus, hepatitis B) should be brought up-to-date. However, live vaccinations are contraindicated in patients on immunosuppressive therapies.
The main objectives of therapy are to heal existing lesions, prevent the formation of new blisters and improve the patient’s quality of life. The goal of management is to induce and maintain remission with the lowest possible doses of medication, so as to minimize the risk of serious and potentially life-threatening drug-related adverse events. Though the optimal therapeutic strategy for PV patients has yet to be established, systemic corticosteroids remain the cornerstone of treatment.
Therapeutic Interventions
Systemic corticosteroids and immunosuppressive therapies are the mainstay of treatment in PV patients. Given the rarity of this disease, studies are limited by small sample size, varied methodologies and a lack of standardized outcomes. As a result, there is a paucity of high-quality, randomized controlled trials (RCTs). Herein, we present a thorough review on the safety and efficacy of the therapeutic interventions for PV (Table 1.1).
Table 1.1
Therapeutic options for the treatment of pemphigus vulgaris
Drug class | Medication | Route | Dose |
|---|---|---|---|
Systemic corticosteroids | Dexamethasone | Oral or IV pulse | 50–200 mg/d for 3–5 d |
Methylprednisolone | IV pulse | 500–1000 mg/d for 3–5 d | |
Prednisone | Oral | 0.5–2 mg/kg/d | |
Immunosuppressive & anti-inflammatory therapies | Azathioprinea | Oral | 0.5–2.5 mg/kg/d |
Cyclophosphamide | Oral | 2–3 mg/kg/d | |
IV pulse | 0.5–1 g/m2 monthly | ||
Immunoablative high-dose IV | 50 mg/kg/d for 4 d | ||
Cyclosporine | Oral | 2–5 mg/kg/d | |
Dapsone | Oral | 25–200 mg/d | |
Gold | IM | 25–50 mg/biweekly | |
Oral | 6–9 mg/d | ||
Methotrexate | Oral or SC | 10–25 mg/week | |
Mycophenolate mofetil | Oral | 2–3 g/d | |
Pentoxifylline | Oral | 1500 mg/d | |
Sulfasalazine | Oral | 1200 mg/d | |
Biologics | Etanercept | SC | 50 mg weekly |
Infliximab | IV | 5 mg/kg/cycle | |
IVIGb | IV | 1–2 g/kg/cycle | |
Rituximab | IV | 375 mg/m2 weekly for 4 weeks or 1000 mg on days 1 and 15 |
Topical Therapies
Baths containing antiseptics (i.e., chlorhexidine) are often recommended to reduce the risks of secondary infections in patients with extensive skin involvement [11]. Potent topical corticosteroids (i.e., clobetasol proprionate 0.05 %) and calcineurin inhibitors (i.e., tacrolimus 0.1 % ointment and pimecrolimus 1 % cream) are beneficial in the treatment of localized skin and/or mucous membrane lesions [13–15]. For vegetative plaques, intralesional corticosteroids (i.e., triamcinolone acetonide 2.5–10 mg/ml) may provide symptomatic relief [13]. Oral topical formulations (i.e., triamcinolone acetonide 0.1 % paste) and inhaled corticosteroids (i.e., mometasone furoate monohydrate nasal spray) can also be used to enhance the delivery of corticosteroids to mucosal surfaces. The use of dental trays improves the efficacy of topical therapies for lesions affecting the gumlines and hard palate. Topical epidermal growth factor (EGF) has also been shown to hasten the healing of PV lesions [16].
Nicotine, a cholinergic agonist, has been proven to induce T cell anergy and improve antibody-mediated acantholysis in pemphigus [17, 18]. Further, remission may be achieved sooner in smokers than in non-smokers [19]. Given the many harmful effects caused by smoking, cigarette smoking is not recommended to PV patients. However, 4 % pilocarpine gel, a cholinomimetic, has been shown to improve the lesional rates of re-epithelialization in PV patients [20].
Systemic Glucocorticoids
Systemic steroids remain the first-line treatment for PV patients. Though the dose of prednisone has been the subject of debate, Ratnam’s RCT suggests that low-dose (i.e., 45–60 mg/day) is as effective as high-dose (i.e., 120–180 mg/day) prednisone [21]. This study, which included 22 participants, did not demonstrate a difference in any of the reported outcomes, including disease control, relapse and death. An RCT of 20 patients further demonstrated that adjuvant pulsed oral dexamethasone treatments provided no additional benefit to conventional first-line therapies [22]. Moreover, significant adverse events occurred more commonly in the pulsed steroid group.
Most expert opinions suggest the initial use of prednisone 1 mg/kg/day, though ranges in dose from 0.5 to 1.5 mg/kg/day have been proposed [13, 23, 24]. If control is not obtained within 2 weeks, a higher dose of prednisone (2 mg/kg/day) may be considered [13]. The approach to steroid dosage must be dynamic and adjusted according to disease severity, underlying patient co-morbidities and response to treatment.
Patients treated with systemic corticosteroids should receive calcium and vitamin D supplementation. To prevent steroid-induced osteoporosis, bisphosphonates (i.e., alendronate, risendronate) should be considered in at-risk patients [25]. The prophylactic use of H2-blockers or proton pump inhibitors for steroid-induced peptic ulcers remains controversial, and treatment should be individualized to the patient [26]. Annual ophthalmologic examinations are recommended to screen for steroid-related ocular complications (i.e., cataracts, glaucoma). Systemic antibacterial, antifungal and antiviral therapies are recommended when clinically indicated [13].
Adjuvant Immunomodulatory Therapies
Azathioprine
Azathioprine, a purine analog which inhibits DNA/RNA synthesis, has long been known to have immunosuppressive and anti-inflammatory effects. In RCTs of pemphigus patients, azathioprine has been compared to corticosteroids alone, mycophenolate mofetil (MMF), cyclophosphamide and tacrolimus [24–31]. Chams-Davatchi et al. conducted a landmark, multi-arm RCT (n = 120) which compared prednisolone alone to three adjuvant therapies: MMF, azathioprine and pulsed cyclophosphamide [27]. Azathioprine demonstrated a steroid-sparing effect when compared to prednisolone alone [27, 28]. Though MMF appeared more effective than azathioprine at achieving disease control, azathioprine showed superior steroid-sparing properties [27, 30]. When compared to cyclophosphamide, azathioprine again demonstrated superior steroid-sparing effects; however, its effect on disease control was inconclusive [27, 29]. In the only trial comparing azathioprine to tacrolimus, there was no significant benefit in any outcome measures [31]. In a meta-analysis of PV therapies, azathioprine was considered to be a steroid-sparing treatment option that had no effect on remission, relapse rate or death [32].
The functional enzyme assay for TPMT should guide physicians when dosing azathioprine. Genetic polymorphisms have been associated with high, intermediate, low and very low levels of TPMT activity. The awareness of these phenotypes allows the physician to: (1) minimize the risk of myelosuppression, and (2) optimally dose azathioprine according to the patient’s TPMT level [33, 34]. Azathioprine should be dosed in accordance with the following recommendations:
Very low TPMT levels (i.e., <5.0 U): azathioprine contraindicated;
Low TPMT levels (i.e., between 5.0 and 13.7 U): up to 0.5 mg/kg/day;
Intermediate TPMT levels (i.e., between 13.7 and 19.0 U): up to 1.5 mg/kg/day;
High TPMT levels (i.e., >19.0 U): up to 2.5 mg/kg/day.
If TPMT functional assays are not routinely available, patients should be started on low-dose (i.e., 50 mg/day) azathioprine. The dose should be slowly increased and blood counts must be closely monitored throughout.
Cyclophosphamide
A derivative of nitrogen mustard, cyclophosphamide is an alkylating agent that acts by cross-linking DNA. Cyclophosphamide preferentially targets B over T lymphocytes, and has potent immunosuppressive properties. Studies have compared cyclophosphamide to corticosteroids alone, azathioprine, MMF and cyclosporine [27, 29, 35, 36]. As a steroid-sparing treatment, cyclophosphamide appears to be more effective than MMF, but less effective than azathioprine [27]. There were no significant benefits for patients receiving pulse dexamethasone-cyclophosphamide as compared to oral methylprednisolone-azathioprine therapy [29]. In a randomized trial to assess the efficacy of adjuvant pulse intravenous cyclophosphamide therapy, there were trends towards fewer relapses and reduced times to remission in the cyclophosphamide-treated group [36]. However, in the meta-analysis by Atzmony et al., cyclophosphamide was found to have no effect on remission rate, relapse rate or time-to-disease control [32]. Though one PV patient died of sepsis during cyclophosphamide therapy, the overall rates of withdrawal due to adverse events were similar among the various treatment groups [32, 36].
The long-term use of cyclophosphamide is associated with significant adverse effects, including infertility, carcinogenicity and hemorrhagic cystitis. Given the lack of evidence and the potential for drug-related toxicities, cyclophosphamide should be reserved for cases that have failed conventional therapies. When treatment with cyclophosphamide is indicated, it may be administered intravenously (i.e., 0.5–1.0 g/m2) or orally (i.e., 2 mg/kg/day) [13]. Few case reports suggest a potential role for immunoablative high-dose cyclophosphamide without stem cell rescue [37].
Cyclosporine
Cyclosporine, a calcineurin inhibitor, is a potent immunosuppressant widely used in transplantation to prevent organ rejection. The efficacy of cyclosporine in the treatment of PV has been evaluated in two small RCTs [35, 38]. As compared to steroids (i.e., prednisone, methylprednisolone) or cyclophosphamide, cyclosporine offers no significant benefit in terms of cumulative corticosteroid dose, disease control, remission, or relapse rates. Adverse events were similar in all treatment groups. Given the lack of evidence, cyclosporine is not routinely recommended in the treatment of PV.
Dapsone
Dapsone, an antibiotic commonly used in the treatment of leprosy, has potent anti-inflammatory and immunomodulatory properties. The efficacy of dapsone has been studied in one small RCT of 19 pemphigus patients who were unable to taper their dose of prednisone below 15 mg/day [39]. While a trend towards efficacy of dapsone was demonstrated, the study was underpowered and the data were inconclusive. There was no significant effect of dapsone on the rate of remission. No patients withdrew from the study because of adverse events. In patients with a normal G6PD level, dapsone is typically dosed between 100 and 150 mg daily.
Intravenous Immunoglobulin
Derived from purified human plasma, intravenous immunoglobulin (IVIG) contains supraphysiologic levels of IgG as well as traces of other immunoglobulins. It exerts a variety of immunomodulatory effects and has been used in the treatment of immunodeficiencies, autoimmune disorders and infections. Though multiple case series suggest a role for IVIG in the treatment of PV, there is only one RCT which evaluates its efficacy in pemphigus patients [40]. A total of 61 PV or PF patients, resistant to treatment with prednisolone, were randomized to receive a single course of adjuvant IVIG (400, 200 or 0 mg/kg/day) for 5 consecutive days. In the three groups, a dose-response relationship was observed. The duration of response, as measured by time to escape from protocol, was significantly improved in the 400 mg group as compared to placebo. There was a significant reduction in the pemphigus activity scores and the ELISA levels in the 400 mg group. No significant differences in safety end points were noted.
Due to the risk of fluid overload, congestive heart failure and renal failure are relative contraindications to the use of IVIG. Patients with IgA deficiency and those with hypercoagulable states are at increased risk for anaphylaxis and thromboembolic events, respectively. Further, treatment with IVIG may predispose patients with rheumatoid arthritis and cryoglobulinemia to renal disease. Screening bloodwork prior to initiating treatment with IVIG includes: a complete blood count, liver and renal panels, IgA level, rheumatoid factor, cryoglobulins, as well as hepatitis B, hepatitis C and HIV serologies. The typical dosing regimen for IVIG is 2 g/kg/month, with the infusions divided over 2–5 days [41, 42].
Methotrexate
Methotrexate, a folic acid antagonist, has anti-inflammatory and immunosuppressive effects. Though methotrexate has been used as a treatment for PV since the 1960s, there are no RCTs evaluating its efficacy. Case series support the use of methotrexate as a steroid-sparing option for PV [43–45]. In the most recent retrospective chart review of 23 PV patients, Tran et al. demonstrated a steroid-sparing effect for methotrexate [46]. Sixteen patients (70 %) were able to taper and ultimately discontinue prednisone within a median time of 18 months. An additional 23 % of patients demonstrated a partial steroid-sparing effect. Methotrexate was only discontinued in two patients (9 %) due to adverse events.
Methotrexate is typically dosed between 10 and 25 mg/week, and may be administered orally or subcutaneously. The use of folic acid supplementation to reduce gastrointestinal adverse effects and pancytopenia remains controversial.
Mycophenolate Mofetil
Mycophenolate mofetil (MMF) is a non-competitive inhibitor of inosine monophosphate dehydrogenase, an important enzyme in the de novo purine synthesis pathway. As this pathway is the major route of purine synthesis for T- and B-lymphocytes, MMF has potent immunosuppressive properties. Studies have compared MMF to corticosteroids alone, azathioprine and cyclophosphamide [27, 30, 47, 48]. As compared to corticosteroids alone, MMF demonstrated a faster and more durable response [47]. Though Ioannides et al. found no significant difference in relapse rate, another study suggested that the time to relapse was delayed in patients treated with MMF [47, 48]. Further, MMF-treated PV patients demonstrated a complete response more rapidly than azathioprine-treated patients [30]. In terms of steroid-sparing effects, MMF showed inferiority to both azathioprine and cyclophosphamide [27, 30]. There were no significant differences noted between the study groups on remission, death or withdrawals due to adverse events.
The typical dosing regimen for MMF is between 2 and 3 g/day. Though the efficacy of enteric coated-mycophenolate sodium in PV patients has yet to be established, it may prove useful in patients presenting with MMF-induced gastrointestinal symptoms.
Plasmapheresis, Immunoadsorption and Extracorporeal Photopheresis
Plasmapheresis has been used to treat a variety of autoimmune disorders that require the rapid removal of disease-causing autoantibodies from the circulation. Guillaume et al. studied the role of plasmapheresis in a multicenter randomized trial of 40 pemphigus patients [49]. As compared to prednisolone alone, the plasmapheresis/prednisolone group showed no significant improvement in disease control, cumulative steroid dose or serum antibody titers. While there was no significant difference in mortality rates, 4 of 22 patients in the intervention group died of thromboembolism or infection.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


