Fig. 2.1
(a) Typical pruritic, eroded and crusted plaques. (b) Typical pruritic, crusted plaques on the chest in a seborrheic distribution. (c) Erosions and erythematous plaques on the back of a woman with pemphigus foliaceus
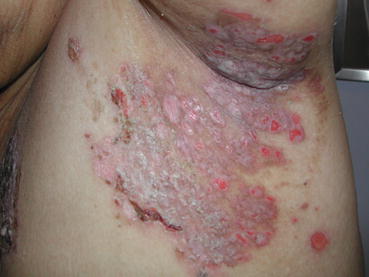
Fig. 2.2
Well-demarcated eroded plaques in the axilla of a patient with pemphigus foliaceus
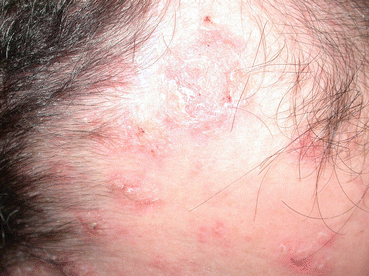
Fig. 2.3
Hyperkeratotic, slightly eroded plaques on the forehead, temple and hairline of a patient with pemphigus foliaceus
Fig. 2.4
Scalp involvement with pemphigus foliaceus
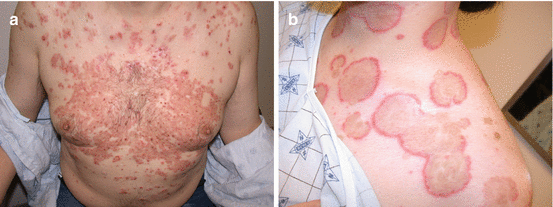
Fig. 2.5
(a) Confluence of erythematous plaques with small erosions in a patient with pemphigus foliaceus. (b) Annular, circinate and geographic plaques with peripheral erosions in this morphological variant of pemphigus foliaceus
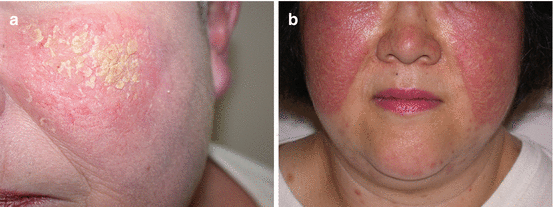
Fig. 2.6
(a) Hyperkeratotic plaque with subtle surface erosions in a patient with pemphigus erythematosus. (b) Bilateral erythematous plaques with light non-confluent scale over the malar cheeks in a patient with pemphigus herpetiformis
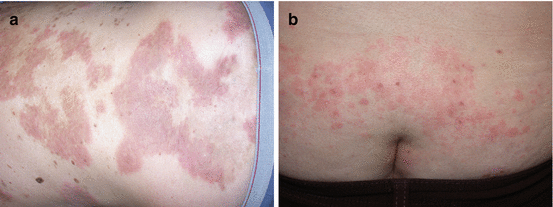
Fig. 2.7
(a) Indurated confluent plaques with an urticarial quality over the back in a patient with pemphigus herpetiformis variant. (b) Urticarial plaques over the gluteal cleft with small vesicles in a patient with pemphigus herpetiformis variant
Demographics
In Europe and the USA the sporadic form is most common with an incidence of about a fifth to a tenth that of PV. The susceptibility genes associated are HLA DRB1*0102 and 0404 [3]. The average age of onset is between 40 and 60 years, has an equal gender distribution and can affect all races and ethnicities [1].
The endemic form (also known as fogo selvagem and Brazilian PF) is most often diagnosed in certain parts of Brazil, Tunisia and Colombia, typically arising at the interface between developing and non-developed areas. Younger individuals in their teens and twenties and multiple members of the same family can be affected. Epidemiological studies suggest an environmental cause which is as yet unidentified, although an insect vector is highly suspected [4]. It seems that as an area becomes more urbanized the disease disappears [5]. The associated susceptibility genes are HLA DRB1*0102, 0404, 1402, and 1406 [6]. Clinically and histologically, both forms of the disease are similar. Globally the incidence and prevalence of PF is very low, however due to the endemic variant and depending on the geographic area being studied, figures may vary significantly.
Diagnosis
Diagnosis is based on a combination of clinical features, histopathological findings and the presence of autoantibodies on direct immunofluorescence (DIF) and indirect immunofluorescence (IIF). ELISA studies can also be sought. None of these tests are exclusively diagnostic and the diagnosis of PF is based on a combination of the above along with a strong clinical suspicion. Histology and DIF require skin biopsies. The biopsy for DIF should be perilesional, normal-appearing skin adjacent to a representative lesion. IIF and ELISA require serum samples.
Histopathology
A superficial bulla with the split high in the granular layer or directly beneath the stratum corneum is typical of an established PF lesion [1]. The bullae contains fibrin, neutrophils, and scattered acantholytic keratinocytes [7]. Early findings include the formation of vacuoles in the intercellular spaces in the upper layers of the epidermis [8]. These can expand, leading to cleft formation. Eosinophilic spongiosis can be seen as a precursor lesion, or as the prominent lesion in the PH variant [9]. Neutrophilic spongiosis is a rare occurrence in PF [10, 11]; and is usually related to the deposition of IgA. Neutrophilic pustules are also an uncommon finding [12]. Later findings include subcorneal blisters in the upper epidermis, which may contain neutrophils and fibrin. The epidermis may be hyperplastic, with overlying focal parakeratosis and some orthokeratosis [13]. Dyskeratotic cells with hyperchromatic nuclei reminiscent of the ‘grains’ found in Darier’s disease can sometimes be found in the granular layer [14]. The superficial dermis is edematous with a mixed inflammatory cell infiltrate (eosinophils and neutrophils predominantly) [13]. Follicular plugging may be present and is important to note for if little else is evident but a clinical suspicion of PF exists [1].
Direct Immunofluorescence (DIF)
The DIF staining shows an intercellular pattern positive for IgG and C3 in both affected and normal skin [15]. This gives a “chicken wire” appearance to the epidermis. The intensity is generally greater in the upper epidermis and sometimes is localized to this level [16]. PE may stain both the intercellular spaces and the basement membrane zone if there is true overlap with systemic lupus erythematosus [17]. Most commonly, PE simply demonstrates the findings of PF (IgG and C3 intercellularly) and appears confined to a clinical distribution seen in acute LE.
IIF Indirect Immunofluorescence (IIF)
IIF uses patient serum to identify antibodies directed against an antigen on a specific substrate. IIF demonstrates circulating antibodies in nearly 90 % of cases of non-endemic PF depending on the substrate utilized [18]. Human skin has a greater density of dsg-1 and therefore has been found to be more sensitive than monkey or guinea-pig esophagus for PF diagnosis [19, 20]. The use of both however increases sensitivity to close to 100 % [21]. Similar to DIF, IIF will show intercellular staining with most fluorescence in the upper epidermis [22]. IIF titers can be used to assess disease activity [23]. Higher titres generally depict more severe disease and vice versa.
Enzyme-Linked Immunosorbent Assay (ELISA)
ELISA uses purified recombinant human dsg-1 to detect PF IgG autoantibodies in patient serum [24]. ELISA also provides a method of measuring the amount of circulating antibodies and hence, can be useful in monitoring a response to treatment [25]. Unfortunately, ELISA and IIF both, are not restricted to the detection of only pathogenic antibody, and will detect any circulating antibodies present to dsg-1.
Systemic Treatment
Dapsone
Dapsone is a sulfone derived antibacterial agent initially used for the treatment of leprosy [26]. It is predominantly used in dermatological conditions with an abnormal accumulation of myeloid cells as it inhibits neutrophil and eosinophil activation and recruitment [27]. It is also used in some autoimmune bullous disorders (AIBD), however its mechanism of action in antibody-mediated diseases e.g. PF remains unclear. As one of the earliest changes observed in PF can be eosinophilic spongiosis, dapsone may be advantageous in the therapeutic armentarium. Adverse effects particularly in patients who are glucose-6-phosphate dehydrogenase (G-6PD) deficient (relative or functional) include methaemoglobinemia and anemia. More rare and serious side-effects can include agranulocytosis and dapsone hypersensitivity syndrome [26]. Gurcan and Ahmed [28] performed a retrospective review of 18 PF patients treated with dapsone. This included a case series of 9 patients reported by Basset et al. [29] and a remaining 9 individual case reports. Overall, 14 of 18 patients responded to dapsone alone (dose-range 100–300 mg/day) or in combination with prednisolone (dose-range 25–75 mg/day). Four patients did not respond. A total of 6-patients developed adverse effects, in 2 this necessitated discontinuation of treatment [28]. Several cases of pediatric sporadic PF including 3 in the Gurcan and Ahmed [28] review responsive to dapsone have been reported [30, 31]. Similarly, the variant PH with heavy eosinophilic spongiosis appears to be very dapsone-responsive [32].
Methotrexate
Methotrexate (MTX) is a folate antagonist initially used in the treatment of malignant disease and later used as an immunosuppressive agent [33]. MTX was one of the first immunosuppressive agents to be used in AIBD. High doses, up to 150 mg weekly, were used leading to severe side-effects [34]. Subsequently MTX use fell out of favour but has been re-emerging more recently. Gurcan and Ahmed [35] analyzed the data to date regarding MTX and PF. In three studies [36–38], a combination of systemic corticosteroids and MTX were used. Twenty patients were treated in total, with a dose range between 12.5 and 37.5 mg weekly. Fifteen patients showed clinical improvement when treated with 20–37.5 mg weekly for 1–27 weeks. At the time of reporting all patients were still on MTX. Four deaths were recorded in this group (3 bronchopneumonia, 1 cerebral thrombosis). Five patients did not improve on MTX. The dosages at the conclusion of MTX were not provided so a corticosteroid-sparing effect could not be determined.
Azathioprine
Azathioprine is a purine analog and prodrug of 6-mercaptopurine. It is metabolized to 6-thioguanine and can block purine synthesis, thus inhibiting rapidly-dividing cells in both de novo and salvage pathways, causing immunosuppression. Testing for normal thiopurine methyl transferase (TPMT) levels can minimize potential bone marrow toxicity with this drug. There are no studies examining the effect of azathioprine in PF alone. Any existing literature is for both PV and PF. Beissert et al. [39] examined 38 pemphigus patients (7 PF). They found no significant difference between azathioprine or mycophenolate mofetil (MMF) both in combination with corticosteroids. However, they subsequently scrutinized this in relation to remission of disease and corticosteroid sparing effects. The patients who were treated with azathioprine received a mean steroid dose of 8.916 ± 29.844 mg. The mean duration of follow-up was 438-days. The duration to disease control in 50 % of patients was less in the azathioprine treated patients as compared to the MMF group (30 vs. 75 days). After 200-days of treatment the remission rate was 75 % in the azathioprine group.
Rose et al. [40] compared dexamethasone/cyclophosphamide pulse therapy with oral methylprednisolone/azathioprine therapy in 22 patients (6 PF). Two patients with PF were examined in the methylprednisolone/azathioprine group. A trend was found in favour of methylprednisolone/azathioprine for remissions and for dexamethasone/cyclophosphamide for side-effect profile. Overall no significant differences were found regarding safety and efficacy.
van Dijk and van Velde [41] reported a case series of ten patients with either pemphigus or pemphigoid treated with azathioprine. Two patients with PF were graded as having a very good response to treatment without severe side-effects. Roenigk and Deodhar [42] also reported a case series of pemphigus patients (1 PF) treated with azathioprine. The PF patient had an excellent response with no side-effects reported.
Mycophenolate Mofetil
MMF is the 2-morpholinoethyl ester of mycophenolic acid. By inhibiting inosine monophosphate dehydrogenase in the de novo purine synthesis pathway, it has the ability to inhibit T and B-cell proliferation, induce T-cell apoptosis and inhibit B-cell antibody production [43–46]. In a study to investigate the safety and efficacy of oral methylprednisolone combined with azathioprine or MMF for the treatment of pemphigus, Beissert et al. [39] assigned 21 pemphigus patients to the MMF group (17 PV, 4 PF). The dose regimen was MMF 2 g/day plus methylprednisolone 2 mg/kg/day, the mean duration of follow-up was 438-days. Complete healing of the lesions and disease remission was noted in 20 (95 %) of 21 MMF patients. One patient (5 %) was noncompliant, discontinued treatment prematurely and did not achieve remission. Complete remission was seen after a mean of 91 ± 113 days. The mean disease-free interval from the time when complete remission was achieved until recurrence of lesions was 123 ± 103 days. Overall it was found that both immunosuppressants (MMF and azathioprine) had a similar efficacy and safety.
Powell et al. [47] described their experience of the addition of MMF to prednisolone in the management of 17 severe refractory pemphigus patients (12 PV, 4 PF, 1 paraneoplastic pemphigus). The regimen used was MMF 750 mg–3.5 g/day plus prednisolone 15–60 mg/day. At the time of reporting only one of the PF cases had clinically inactive disease. Two cases have experienced improved disease control and one case has clinically active disease responding recently to the addition of monthly-pulsed intravenous immunoglobulin 2 mg/kg/month to a combination of prednisolone 20 mg/day and MMF 3 g/day. The average doses of prednisolone prior to commencing MMF therapy and currently for this group are 35 mg and 8 mg daily, respectively.
Mimouni et al. [48] examined 42 patients with pemphigus (31 PV, 11 PF) who had relapses during prednisolone taper or were unable to tolerate side effects from previously used drugs. With MMF, complete remission was achieved in 22 PV and 5 PF patients and partial remission in 1 PV and 4 PF patients. Two treatment failures were recorded in the PF group. The median treatment/follow-up period was 22-months (range: 4–49 months) and the median time to remission was 9-months (range: 1–13 months). There was no statistical difference in the number of complete/partial remission or adverse effects between PV and PF patients.
Several other isolated cases of PF treated successfully with MMF are reported in the literature. Nousari et al. [49] reported a case of a 55-year-old male with only partial response to prednisolone and hepatoxicity to azathioprine. MMF was commenced at a dose of 1 g/twice daily, progressive improvement within 5-weeks of therapy was noted which allowed a gradual taper of prednisolone. At 6-months he was in complete remission receiving prednisolone 10 mg/day and MMF 1 g twice daily with no reported side-effects. Katz et al. [50] reported a 37-year-old female with PF initially treated with doses of prednisolone up to 60 mg/day and azathioprine 250 mg/day resulting only in partial remission. She was switched to MMF 1 g twice daily and prednisolone was continued at 40 mg/day. The patient improved over a 6-week period but flared when prednisolone was tapered below 20 mg/day. MMF was increased to 1.5 g twice daily and prednisolone was successfully tapered to 7.5 mg/day. At 9-months the patient remained clear with no flares.
Bongiorno et al. [51] investigated the efficacy of enteric-coated mycophenolate sodium (EC-MPS) 1440 mg daily and prednisolone 75 mg daily, in ten patients with active, refractory PV or PF over 18-months. A single PF patient was included, at 18-months had clinically quiescent disease. By 6-months the EC-MPS dose had been reduced to 720 mg/day and prednisolone 15 mg/day, this regimen continued at 18-months.
Intravenous Immunoglobulin
Intravenous immunoglobulin (IVIg) is made from IgG fractionated from pooled plasma via whole blood donors or by plasmapheresis and is usually administered in a monthly dose [52]. IVIg blocks fetal Fc receptors resulting in catabolism of circulating antibodies. As IVIg does not contain pemphigus antibodies, the patient ends up preferentially breaking down the pathogenic pemphigus antibodies as normal antibodies are being replaced. To prevent rebound auto-antibody production when fetal Fc receptors again become exposed, IVIg is often used with a systemic immunosuppressive agent capable of suppressing antibody production. Generally the use of IVIg is restricted to patients who: fail conventional therapy; have side-effects or contraindications to conventional therapy; and/or have rapidly progressive disease; have a very high indirect antibody titer [53]. The majority of the reported cases of IVIg use in pemphigus utilize the 2 g/kg/cycle dose [54]. Amagai et al. [55] in a randomized, double-blinded, placebo-controlled trial investigated the therapeutic effect of a single cycle of IVIg compared to conventional therapy with systemic corticosteroids in 61 patients with either PV or PF. If pemphigus lesions improved, patients were allowed to stay on protocol; patients not responding to therapy were removed from the study. Those randomized to receive IVIg, stayed in the study significantly longer, showed clinical improvement and suppression of autoantibody levels, compared to those randomized to conventional therapy. The follow up period was 90-days. The authors concluded that a single cycle of IVIg is rapidly effective in the treatment of pemphigus. However, most clinicians utilize multiple courses of IVIg to maintain disease control. In their consensus statement on IVIg use, Ahmed and Dahl [56] proposed its use at a dose of 2 g/kg divided over 3–5 days every 4-weeks until disease control is obtained. They recommend the interval between infusions is then slowly increased to 6, 8, 10, 12, 14, and 16-weeks and then stopped. If the disease flares, the frequency of infusions is increased until control is obtained and then the tapering regimen is again resumed. Jolles [57] reported that AIBD seem to have a better response to IVIg when used concomitantly with other treatments. IVIg in combination with systemic corticosteroids and/or immunosuppressive agents exhibited 91 % response rate compared to 56 % response rate when used as monotherapy, likely due to increased synthesis or rebound of pathogenic antibody. The addition of cytotoxic agents e.g. cyclophosphamide appears to make IVIg even more effective, as this agent is one of the strongest suppressors of antibody production from B-cells [52].
Cyclophosphamide
Cyclophosphamide is a nitrogen mustard with potent effects upon B-cells and strong suppression of antibody production. Pasricha et al. [58] first described the use of high-dose dexamethasone and cyclophosphamide pulse therapy (DCP) for pemphigus patients. The treatment regimen was divided into four-phases. Phase I consisted of monthly 100 mg intravenous (I.V.) dexamethasone on 3-consecutive days and 500 mg of intravenous cyclophosphamide on one of the days plus daily oral cyclophosphamide (50 mg). This was repeated until clinical remission was achieved (phase I) and then a further 6-months of DCP treatment was given (phase II). If remission was maintained DCP treatments were stopped and oral cyclophosphamide was continued for a further year (phase III); all treatment was withdrawn thereafter if patients remained in remission. Pasricha et al.’s [58–64] data suggested that this regimen was highly effective in inducing remission with minimal toxicity.
Between 1982 and 1998, 500 patients (PV 444, PF 33, PE 18, and pemphigus vegetans 5), with an almost equal sex ratio (251 males, 249 females) were enrolled for DCP regimen. The patient ages varied; 44 patients <20 years, 246 patients between 20 and 40 years, 190 patients between 40 and 60 years and 20 >60 years. Of these, 97 patients could not complete treatment, 19 patients died due to a variety of causes, most of which were unrelated to the disease or its treatment, or causes that were preventable with better patient management. The remaining 384 patients recovered from the disease and at the time of reporting were living without any disease and without any maintenance treatment [64]. Pasricha et al. conclude that pemphigus can be controlled in almost every patient and if a patient strictly follows the DCP regimen, a cure can be achieved.
In a multicenter, prospectively randomized study, Rose et al. [40] compared efficacy and side-effects of DCP with a methylprednisolone-azathioprine (M/A) therapy in 22 pemphigus patients, 6 of whom had PF. Eleven received DCP. This consisted of 100 mg I.V. dexamethasone per day on 3 consecutive days with cyclophosphamide (500 mg) day 1. Initially pulses were repeated every 2–4 weeks and then at increasing intervals. In between pulses, oral cyclophosphamide (50 mg) was given daily for 6-months. The results showed that within 24-months after treatment initiation, 5 of the 11 patients of the DCP group had a remission (complete remissions after discontinuation of therapy in 3 patients) and 6 of the 11 patients had a progression. There were more relapses in M/A therapy after remission than in DCP therapy. Side-effects were more common in the M/A group. These differences were not significant. The authors concluded that due to the high number of progressions in patients treated with DCP, the encouraging results of earlier reports about DCP could not be confirmed. However DCP was better tolerated and, in the case of primary efficacy, was associated with fewer recurrences than M/A therapy.
Saha et al. [65] presented a retrospective review of 21 patients (2 PF) treated with pulsed cyclophosphamide and high-dose methylprednisolone over a 10-year period. These patients had all been refractory to steroid and adjuvant either azathioprine or MMF. Of the treated patients the responses were: 7 excellent, 2 good, 5 moderate, 6 minimal and 1 had no clinical response. Four patients achieved complete clinical remission and the number of pulses for these patients varied between 11 and 22. All patients were able to reduce their prednisolone dose from a pre-pulsing median dose of 40–10 mg at the last pulse with a median dose reduction of 66 % (p < 0.001). The most common adverse effect was transient lymphopenia (12 patients), nonlife-threatening sepsis (7 patients) and pre-mature ovarian failure (2 patients). The authors concluded that pulsed cyclophosphamide can be an effective treatment for refractory pemphigus but its adverse effects should be considered prior to therapy and closely monitored in patients on treatment. Longer-term risks from cyclophosphamide should also be taken into account including risk for bladder cancer and acute myelogenous leukemia that would not have been found in the short time frame of these studies.
Plasmapheresis
Guillaume et al. [66] reported the results of a multicenter randomized study examining the efficacy of plasmapheresis in 40 pemphigus patients (7 PF). Eighteen patients were treated with prednisolone alone and 22 with prednisolone and plasmapheresis. There was no difference in outcome between the treated group and control patients regarding disease control, cumulative corticosteroid dose or serum antibody titers. Four deaths were documented in the treatment group. It was concluded that plasmapheresis in association with low steroid doses are not effective in the treatment of pemphigus and may even promote sepsis.
Rituximab
Rituximab (RTX) is a chimeric murine/human monoclonal antibody that recognizes the B-lymphocyte surface protein CD20, a transmembrane protein expressed on pre-B to mature B-cells and functions to regulate B-cells early in development [67]. RTX completely destroys this phase of B-lymphocyte growth and results in subsequent decline and depletion of pathogenic antibodies and minimizes pathogenic B-cell presentation of auto-antigens to T-cells. RTX has increasingly been reported to be effective in AIBD [68]. Heelan et al. [69] conducted a retrospective study of 92 patients (PV 84, PF 8) to assess the clinical response of patients with pemphigus to RTX using a modified fixed-dose rheumatoid arthritis protocol (1 g I.V. on days 1 and 15, followed by 500 mg if clinically warranted at 6-month intervals or repeated full dosing). Median time to relapse after the first treatment cycle was 15-months (95 % CI, 10.3–19.7). When comparing time to relapse for PV and PF, there was no statistically significant difference. The PV median time to relapse was 15-months (95 % CI, 8.6–21.4), and the PF median time to relapse was 12-months (1.5–22.5; p = 0.99). All patients experienced improvement. Complete remission rates with or without adjuvant treatment at final follow-up were 89 % (56 patients complete remission without treatment, 26 patients complete remission with adjuvant treatment). No serious infectious adverse events occurred.
Reguiai et al. [70] showed that RTX appeared to be a durable, effective, and well-tolerated treatment for severe pemphigus. This retrospective study included 24 patients with severe pemphigus (9 PV, 4 PF), treated with RTX (n = 13) or systemic corticosteroids alone or combined with immunosuppressants (n = 11 control subjects). Of the 13 patients treated with RTX, 9 achieved complete remission 3-months after 1 RTX cycle. Thereafter, 7 patients (4 with maintenance therapy) relapsed within a mean of 18-months after the last RTX cycle and received 1 or 2 additional RTX cycles. Mean follow-up was 41-months after the first cycle and 28-months after the last. All 13 patients remained in complete remission (5 patients off therapy).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


