Fig. 25.1
(a) Schematic of construction of the scFv antibody library and isolation of monoclonal antibodies (mAbs) with specific binding to desmoglein (Dsg) using phage display. (b) Direct disruption of Dsg interactions by pemphigus autoantibodies (steric hindrance). AK23, a pathogenic mAb, binds to the transadhesive interface between Dsg molecules (A) and other mAbs, such as PVA224, PVB28, and PVB124, and reacts with cis-elements between neighboring Dsg molecules (B) [5]
From an adoptive transfer mouse model of pemphigus (details are in the latter part of this chapter), a pathogenic mAb against Dsg3 has been isolated using a cell fusion method [4]. Epitope analysis using Dsg1/Dsg3 domain-swapped molecules and point-mutated Dsg3 molecules revealed that the pathogenic mAb recognized the amino terminal extracellular domain of Dsg3 that is predicted to form the intercellular transadhesive interface, and nonpathogenic mAbs recognized the membrane proximal extracellular domain of Dsg3 (Fig. 25.1b). Another study isolated anti-Dsg3 mAbs using B cells immortalized by Epstein–Barr viral infection derived from the peripheral blood of a PV patient [5]. One of the pathogenic mAbs also bound to the amino terminal extracellular domain of Dsg3. Epitope mapping analysis using peptide libraries revealed that this pathogenic mAb bound to the part associated with cis-binding between neighboring Dsg3 molecules, predicted by positioning of homologous peptides in the C-cadherin crystal structure [6]. Results from a competition enzyme-linked immunosorbent assay (ELISA) using PV sera and this mAb suggested that the epitope can be shared with most sera of PV patients in the study (Fig. 25.1b).
Unlike many other autoantibody-mediated diseases, such as pemphigoid, in which the constant regions of antibodies are required for blister formation to activate complement, or bind FC receptors on inflammatory cells, the variable regions of antibodies are sufficient to cause blisters in pemphigus. Based on cadherin ultrastructure (steric hindrance) and epitope mapping studies using mAbs, several reports have suggested that pathogenic PV and PF autoantibodies bind to calcium-sensitive, conformational epitopes in the amino terminal extracellular domains that form the key molecular interactions for Dsg intercellular adhesion [4, 7].
25.2.2 Common Sequences of CDR3 Region of Pathogenic Monoclonal Antibodies in Pemphigus and Their Binding Target on Hot Spot of Desmogleins
Several studies have been able to isolate mAbs against Dsgs. Inasmuch as mAbs are divided into two groups, pathogenic mAbs and nonpathogenic mAbs, there should be diversity in pathogenicity among autoantibodies in an in vivo situation [8]. It is sometimes observed that an ELISA titer is not necessarily associated with disease severities while following patients with pemphigus. As mentioned here previously, one of the factors determining pathogenicity is the epitope, or the part of the antigen where antibodies are binding. Further characterization of the structure of pathogenic mAbs will probably aid in the discovery of a promising target for a novel therapy in the future.
A series of mAbs against Dsg1 and Dsg3 in the form of scFv have been cloned from patients with PV and PF. Epitopes defined by pathogenic mAbs were shared by sera from various PV and PF patients, suggesting that pathogenic antibodies bind similar sites on Dsg and, therefore, might share common idiotypes. To investigate whether these shared idiotypes might be associated with homologous amino acid sequences in various pathogenic mAbs, variable regions of the mAbs were sequenced. Immunoglobulin has three complementarity determining regions (CDR1, CDR2, and CDR3) in each variable heavy chain and light chain region. Specifically, CDR3 in the heavy chain (H-CDR3), the junction of VH, D, and JH genes, varies the most in structure and plays a crucial role in determining the antigen specificity of an antibody (Fig. 25.2a, b), which is why H-CDR3 was focused on to determine a common motif among pathogenic mAbs. In six out of nine pathogenic clones, a consensus amino acid motif (D/E-X-X-X-W) was found in which an acidic amino acid (aspartic acid [D] or glutamic acid [E]) was arranged for amino acids upstream from a tryptophan (W) [9]. On the other hand, all 21 nonpathogenic mAbs previously cloned did not contain this H-CDR3 consensus motif.
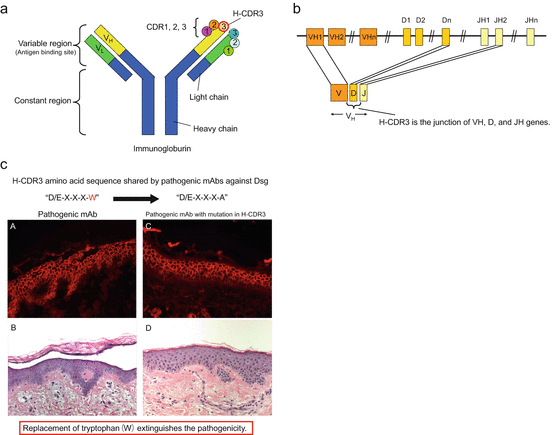
Fig. 25.2
(a, b) Complementarity-determining regions (CDR1, CDR2, and CDR3) in variable regions of the immunoglobulin chain. Specifically, CDR3 in the heavy chain (H-CDR3), the junction of VH, D, and JH genes, plays a crucial role in determining the antigen specificity of an antibody. (c) Pathogenicity impaired by a single amino acid mutation in H-CDR3. A pathogenic mAb had the consensus motif D/E-X-X-X-W in H-CDR3, and the tryptophan (W) was replaced with an alanine (A). The original pathogenic mAb without mutation caused blisters upon injection into human skin organ culture (A, B), whereas the mAb with the W replacement still bound to the epidermis, but lost pathogenicity (C, D) [9]
To determine whether the H-CDR3 sequence must be unique for the binding activity and/or pathogenicity of each mAb, the H-CDR3 sequence of a pathogenic anti-Dsg1 mAb containing the consensus motif was randomized. Nine clones of Dsg1-binding mAbs derived from the mAb with H-CDR3 randomization were obtained. Based on ELISA results, all nine mAbs showed specific binding to Dsg1 and cell surface staining in the epidermis on indirect immunofluorescence using normal human skin. Injection into normal human skin organ culture showed that seven of the nine clones were pathogenic, indicating that a given H-CDR3 imparting pathogenicity to an anti-Dsg mAb was not necessarily unique for each mAb. These results also revealed that antibody binding to Dsgs can be uncoupled from pathogenicity. Although two pathogenic randomized clones shared the consensus motif D/E-X-X-X-W, others did not, suggesting that this consensus motif is not necessary for pathogenicity. However, all randomized clones contained at least one tryptophan (W) in the H-CDR3, suggesting the importance of W for mAbs that bind to Dsg1 and those that cause pathogenicity. To confirm the importance of W in the H-CDR3 of pathogenic mAbs, the W was mutated to an alanine (A) by site-directed mutagenesis. These changes inhibited the pathogenicity of mAbs but not their binding activities, confirming the importance of W for pathogenicity. These findings suggest that W in the H-CDR3 is critical for pathogenicity but not binding, which is in agreement with the randomization studies discussed above (Fig. 25.2c). It is believed that, in desmosomal cadherin, such as Dsg, W is critical in the transinteraction and, thus, antibodies that interfere with binding may make use of a W through inhibition [6].
These observations suggest that autoantibodies have a “hot spot” that determines pathogenicity and, therefore, could be a novel therapeutic target.
25.3 Dsg3-Specific Autoreactive T Cells as a Commander in Chief in Pemphigus
Although the characteristics of pathogenic anti-Dsg3 autoantibodies were discussed above, it is also believed that autoreactive T cells are required to induce autoantibody production. The following evidence supports the commitment of autoreactive CD4+ T cells in the pathogenesis of PV. Strong association of PV with certain HLA class II alleles including DRB1*0402 in Jewish and DQB1*0503 in non-Jewish individuals indicates HLA class II-restricted recognition of Dsg3 by CD4+ T cells [10–14]. Mutations in CDR3 of anti-Dsg3 antibodies suggest CD4+ T cell-dependent somatic hypermutation [1]. However, analysis of human Dsg3-specific T cells is technically difficult because there is no experimental system to elucidate whether analyzed T cells have the capacity to induce anti-Dsg3 antibody production and the PV phenotype in vivo through controlling Dsg3-specific B cells. Over 10 years ago, we reported the experimental mouse model of PV that paved the way for analyzing the pathogenicity of T cells in vivo and gave us new insights into the pathogenesis of pemphigus as well as inflammatory skin diseases [15].
25.3.1 Development of an Active Disease Mouse Model for Pemphigus
It was difficult to make a pemphigus mouse model by repetitive immunization of a wild-type mouse with recombinant Dsg3 protein; this was probably due to strictly regulated immune tolerance against Dsg3. Then, an autoantigen-deficient mouse was utilized to elicit an immune response against the autoantigen, as the deficient mouse does not establish immunological tolerance against the autoantigen. To generate a humoral autoimmune response against Dsg3 that causes pemphigus vulgaris, a Dsg3−/− mouse was immunized with recombinant Dsg3, thus producing an anti-Dsg3 antibody (Fig. 25.3a). Because the Dsg3−/− mouse does not express Dsg3 in the stratified squamous epithelium, the antibody produced does not bind the skin and oral mucosa or induce the PV phenotype. Splenocytes primed with rDsg3 are then transferred to a Rag2−/− mouse , physiologically expressing Dsg3. Transferred splenocytes begin to produce an anti-Dsg3 antibody, which binds to Dsg3 molecules in the skin and oral mucosa. Therefore, the recipient Rag2−/− mouse starts to show the PV phenotype, including weight loss and erosions on the face, where the mouse tends to scratch frequently, followed by gradual spreading to the whole body. Weight loss is believed to be caused by difficulty in eating; specifically, a suprabasilar acantholytic blister, a characteristic feature of PV, is observed in the pathology of the hard palate and esophagus in the recipient (Fig. 25.4a). In addition, IgG deposition on keratinocyte cell surfaces was detected by immunofluorescent staining and anti-Dsg3 IgG titer peaks at 2–3 weeks after transfer, which was confirmed by ELISA (Fig. 25.4a). Unlike PV patients, the PV mouse model showed telogen hair loss caused by Dsg3 dysfunction that is strongly expressed in the follicular epithelium and contributes to cell adhesion during the telogen hair cycle.
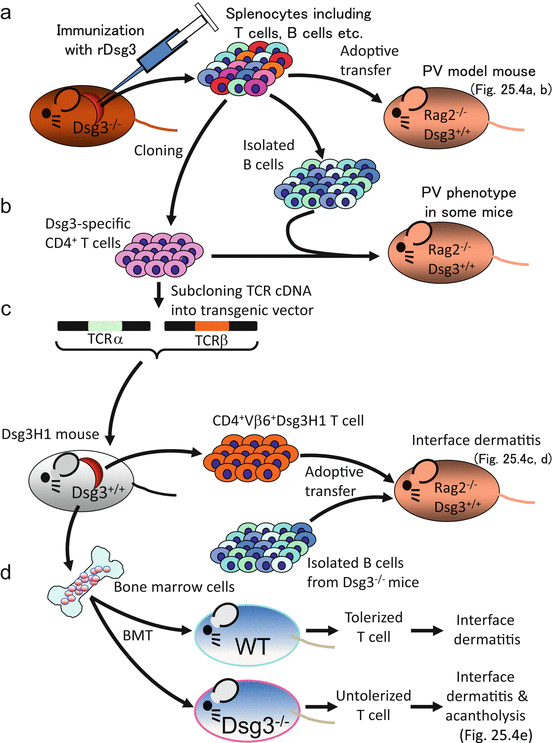
Fig. 25.3




Experimental outline. (a) Approach to make the PV mouse model. (b) Pathogenicity evaluation of Dsg3-specific T-cell clones. (c) Generation of Dsg3H1 mice and pathogenicity evaluation of transgenic T cells. (d) Bone marrow transfer (BMT) experiment and comparison between tolerized and untolerized transgenic T cells
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
