, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
2.1 Ichthyosis Vulgaris
2.1.1 Clinical Features
Ichthyosis vulgaris is a non-syndromic ichthyosis and is the most commonly seen variant of disease with a reported incidence of 1 in 250 to 1 in 1000 individuals [1]. It appears to be exacerbated by cold, dry climates and is seen in association with atopic dermatitis in 50–60 % of the cases.
Ichthyosis vulgaris is characterized by fine, light gray, fish-like scaling, most notably along the lower extremities, sparing the face and skin folds. As previously mentioned, it frequently is seen in the setting of atopy with other clinical findings of xerosis, hyperlinear palms and soles, hypohidrosis, pruritus, and keratosis pilaris [1]. Skin involvement is usually quite mild and may be managed with aggressive use of moisturizers, emollients, and mild keratolytics.
2.1.2 Histology
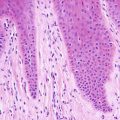
Ichthyosis vulgaris shows orthokeratotic hyperkeratosis and loss of the granular layer [2] (Fig. 2.1). The epidermis is usually of normal thickness, although it may be slightly thinned. A rare variant limited to acral surfaces demonstrates similar epidermal changes associated with the loss of eccrine structures [3]. Acquired ichthyosis associated with HIV, lupus erythematosus, sarcoidosis, and other systemic illnesses has identical histologic features to ichthyosis vulgaris [4].
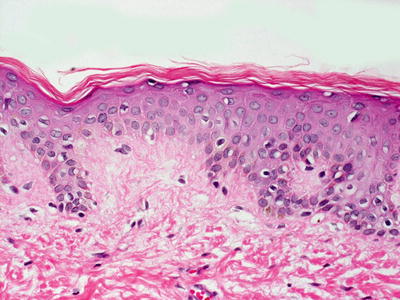
Fig. 2.1
Histologic changes seen in ichthyosis vulgaris include acanthosis and loss of the granular layer with overlying orthokeratotic hyperkeratosis
2.1.3 Pathogenesis
Ichthyosis vulgaris is caused by loss-of-function mutations in the gene filaggrin (FLG) [5–7]. FLG mutations at positions R501X and 2282del4 account for about 80 % of the mutations in northern Europeans [8]. Mutations in FLG gene result in a truncated profilaggrin (filaggrin precursor) protein that cannot be correctly processed into functional filaggrin [9]. Subsequently, filaggrin is significantly reduced or absent in ichthyosis vulgaris [10].
The severity of ichthyosis vulgaris phenotype depends on the extent of loss of filaggrin expression. Patients heterozygous for the mutations have a mild phenotype, whereas those who are homozygous for the mutations exhibit a more severe phenotype [11]. A major characteristic of ichthyosis vulgaris is the absence of the granular layer and abnormal or missing keratohyalin granules [10, 12]. Similar to the mutational genotype and the correlative severity of the disease phenotype, patients with heterozygous mutations in FLG have a reduced granular layer, whereas those with homozygous mutations have a complete loss of the granular layer .
At the molecular level, profilaggrin and filaggrin deficiency has multiple consequences. Profilaggrin is a major protein component of keratohyalin granules in the stratum granulosum [13]. Profilaggrin is proteolytically processed to produce filaggrin, which aggregates keratin filaments to form the cornified cell envelope [13, 14]. Filaggrin protein is essential for epidermal differentiation, lipid metabolism and barrier function [15]. Mature filaggrin is proteolyzed into its constituent amino acids and small peptides with hygroscopic properties that underlie hydration in the stratum corneum [16, 17]. The loss of filaggrin reduces the ability of keratinocytes to remain hydrated, resulting in excessive scale [18]. Defects in filaggrin also cause cytoskeletal disorganization, and impaired organization and maturation of the lamellar bilayers, thus affecting the lipid contents in the stratum corneum. There is also increased permeability of chemicals and allergens in filaggrin-defective skin , leading to increased sensitization to these irritants in ichthyosis vulgaris [15, 19]. Importantly, there is a strong association of ichthyosis vulgaris with atopic dermatitis in the context of FLG null alleles [6, 20].
2.2 Sex-Lined Ichthyosis
2.2.1 Clinical Features
Sex-linked ichthyosis (or X-linked ichthyosis ) is the second most common ichthyosis subtype [21]. It is transmitted in an X-linked recessive manner and affects 1:2000 to 1:6000 males with no racial prevalence [22].
Patients often present with generalized xerosis and scaling with progression to larger, polygonal, adherent, “dirty brown” scales along the extensor surfaces of the extremities and symmetrically along the sides of the trunk. Involvement at the scalp, preauricular cheeks, and neck resolve over childhood (Fig. 2.2) [21]. Comma-shaped corneal opacities are pathognomonic, appearing during adolescence with no apparent effect on visual acuity, although they may predispose affected individuals to corneal erosions. Cryptorchidism is seen in 20 % of individuals with X-linked ichthyosis. The associated deficiency of steroid sulfatase in the fetal placenta may present with failed labor initiation or delayed progression [21, 22].
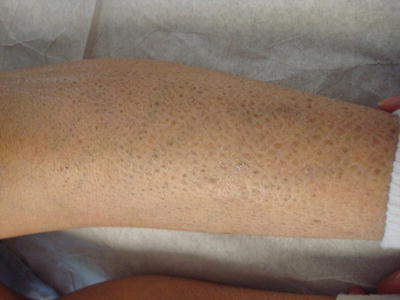
Fig. 2.2
Sex-linked ichthyosis demonstrates brown, adherent and polygonal scales on the lower extremity
In individuals with X-linked ichthyosis, there is an increased risk of testicular germ cell cancer [21]. However, patients generally do very well and follow a benign clinical course with improvement in cutaneous findings over time. Hydration , lubrication, and keratolytics (for thicker areas of scale) are key to disease management.
2.2.2 Histology
Sex-linked ichthyosis shows little histologic change and skin biopsies often resemble normal skin. Some authors have suggested that diagnosis is not possible on biopsy and that enzyme analysis is required to establish the diagnosis [23]. There is slight orthokeratotic hyperkeratosis and the basket-weave pattern is usually preserved (Fig. 2.3). The granular layer is preserved and the epidermis has normal thickness. In one series, a diminished granular layer was reported in some patients with X-linked ichthyosis [2]. Hyperkeratosis overlying eccrine and follicular orifices has been described in some patients with this type of ichthyosis [24]. Some authors believe sex-linked ichthyosis to be identical to steroid sulfatase deficiency and its associated ichthyosis [25, 26]. Others believe the diseases to be similar, but the sex-linked disease to be associated with more severe ichthyosis [27]. On ultrastructural examination, keratohyalin granules are larger and have a spongy appearance [28]. A banding pattern has been described in polarized hair microscopic examination in some of these patients [29].
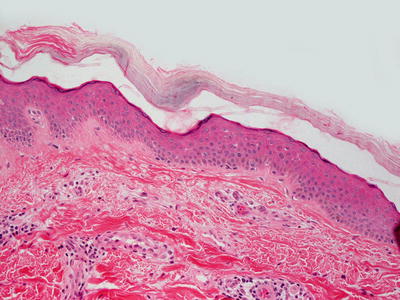
Fig. 2.3
Histologic changes in X-linked ichthyosis include subtle orthokeratotic hyperkeratosis and preserved granular layer. The epidermis usually has normal thickness
2.2.3 Pathogenesis
Sex-linked ichthyosis is a recessive X-linked keratinization disorder that affects males [30]. Female carriers generally do not manifest the disease. Sex-linked ichthyosis is caused by loss-of-function mutations in the steroid sulfatase (STS) gene [31–33]. About 90 % of patients with X-linked ichthyosis have complete or partial deletions of STS [34]. STS is located on chromosome Xp22.3 and encodes an enzyme that is involved in lipid metabolism by catalyzing the hydrolysis of 3-beta-sulfate esters [35]. Loss of STS activity leads to the accumulation of cholesterol sulfate in the stratum corneum, perturbs the cell membrane integrity and increases intercellular cohesion, inducing scale formation [33, 34, 36, 37]. Excess cholesterol sulfate can also lead to separation between corneocytes and impaired skin barrier function in the epidermis. In addition, there is increased calcium in the stratum corneum, which may contribute to corneocyte retention by increasing intercellular cohesion [36].
2.3 Lamellar Ichthyosis
2.3.1 Clinical Features
Autosomal recessive congenital ichthyosis encompasses a wide spectrum of non-syndromic ichthyoses, often presenting with collodion membrane and eventual progression to a number of varied phenotypes including harlequin ichthyosis, lamellar ichthyosis, non-bullous congenital ichthyosiform erythroderma, self-healing collodion baby, acral self-healing collodion baby, and bathing suit ichthyosis [1, 38].
Lamellar ichthyosis is the most common subtype of autosomal recessive congenital ichthyosis with reported prevalence of 1 in 200,000–300,000 neonates [38]. It usually presents at birth with collodion membrane (Fig. 2.4), which is a tight, shiny membrane encasing the entire body with periorificial tightening, causing ectropion and eclabium. Although the membrane is shed within the first few weeks of life, patients later develop generalized, dark-colored, plate-like scales, occasionally with persistence of ectropion. Patients may develop scarring alopecia on the scalp as well as palmoplantar keratoderma (Fig. 2.5).
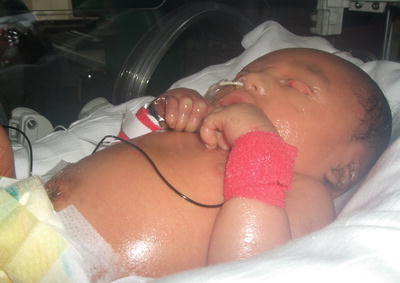
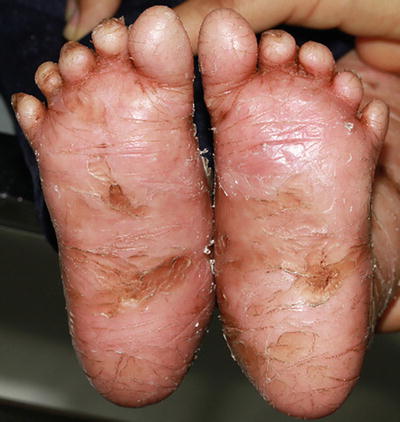
Fig. 2.4
Lamellar ichthyosis baby presents at birth with collodion membrane
Fig. 2.5
Lamellar ichthyosis shows dark-colored scaling in the setting of pronounced keratoderma on the soles of an infant (photo courtesy of Minh Hoang, MD, Ho Chi Minh City, Vietnam)
Disease expression is varied, but most individuals have skin changes that are persistent throughout life [1]. Hyperkeratotic plaques may interfere with sweat gland functioning and predispose patients to hyperthermia [39, 40]. Ectropion places the patient at risk for keratitis and corneal abrasions [39, 40]. Conductive hearing loss may occur secondary to external auditory canal stenosis by cerumen impaction [39]. Collodion membrane has an overall mortality rate of 10–20 % with morbidity secondary to temperature dysregulation, fluid and electrolyte imbalance, infection secondary to impaired skin barrier function and failure to thrive [1]. Management necessitates admission to an intensive care unit.
2.3.2 Histology
Lamellar ichthyosis, renamed ichthyosis congenital type II by some authors [41], is the most prominent variant of ichthyosis congenita and is characterized by compact orthokeratotic hyperkeratosis (Figs. 2.6 and 2.7). Cholesterol clefts are noted within the stratum corneum on electron microscopy.
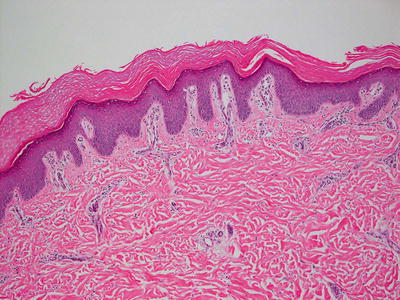
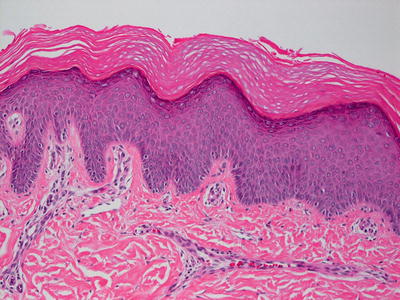
Fig. 2.6
Lamellar ichthyosis is characterized by dense, compact orthokeratotic hyperkeratosis, preservation of the granular layer and an acanthotic epidermis
Fig. 2.7
Hyperkeratosis and hypergranulosis are present in lamellar ichthyosis
Harlequin fetus demonstrates massive, confluent hyperkeratosis. Focal parakeratosis is present in some cases and is accompanied with a diminished granular layer [42]. Large concentric lamellar bodies and giant mitochondria are seen on electron microscopy [43–45]. As there are many histologic similarities between patients with lamellar ichthyosis and those with Harlequin fetus phenotype, electron microscopy is the preferred method in distinguishing between the two entities [46].
2.3.3 Pathogenesis
Lamellar ichthyosis is a type of autosomal recessive congenital ichthyosis that is caused by mutations in the transglutaminase-1 (TGM1) gene [47–50]. Over 50 different mutations in TGM1 have been identified. Some mutations result in disruption of the active site of the enzyme, leading to loss of enzyme activity [11, 51, 52]. Patients with lamellar ichthyosis can be homozygous for single mutations or heterozygous for two different mutations in the same gene.
The function of TGM1 is to crosslink proteins and sphingolipids to the cornified cell envelope [53, 54]. The cornified envelope is an essential scaffold for the intercellular lipid layer formation in the stratum corneum. Defects in TGM1 activity can impair the formation of the lipid layers, leading to defective barrier function and the proliferative hyperkeratosis seen in lamellar ichthyosis. As noted in their clinical presentation, patients with lamellar ichthyosis have increased epidermal turnover and proliferative hyperkeratosis.
As a group of autosomal recessive congenital ichthyoses (ARCI) , several causative genes associated with ARCI have been identified. These genes include transglutaminase 1 , adenosine triphosphate binding cassette 12 (ABCA12) , and the lipoxygenases ALOXE3 and ALOX12B [47, 50, 55–59]. In particular, ABCA12 mutations have been report in lamellar ichthyosis [60]. Other studies, however, have reported ABCA12 mutations in non-bullous congenital ichthyosiform erythroderma, but not in lamellar ichthyosis in Japanese population, noting that these findings may be different among Asian and European populations [61].
2.4 Epidermolytic Hyperkeratosis/Bullous Congenital Ichthyosiform Erythroderma
2.4.1 Clinical Features
Epidermolytic hyperkeratosis (also known as bullous congenital ichthyosiform erythroderma and epidermolytic ichthyosis) is an autosomal dominant disorder with six distinct clinical phenotypes [22]. Patients may present with generalized blistering in the neonatal period, infancy, and early childhood with eventual development of malodorous, corrugated plaques along the flexural folds (Figs. 2.8, 2.9, and 2.10).
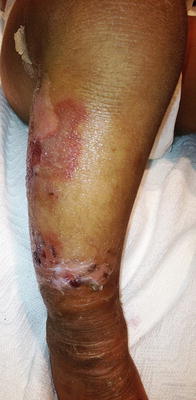
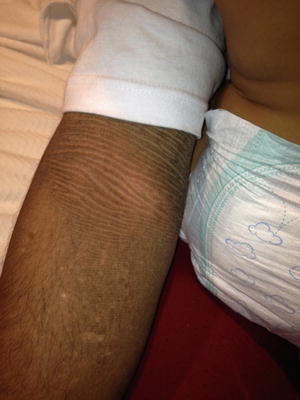
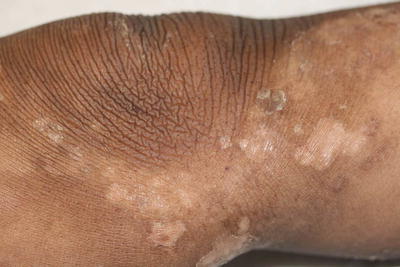
Fig. 2.8
Epidermolytic hyperkeratosis presents with hyperpigmented hyperkeratosis along the extensor surface of the distal lower extremity with multiple erythematous erosions and ruptured bullae
Fig. 2.9
Corrugated plaques along the flexural folds are key features of epidermolytic hyperkeratosis in older patients
Fig. 2.10
Characteristic features of hyperpigmented thickening of the extensor knee with surrounding healing erosions are seen in this example of epidermolytic hyperkeratosis
Skin changes are persistent. As in other ichthyoses, hydration, lubrication and keratolysis are key to disease management [22]. Although the skin appears quite thick, the skin barrier is significantly impaired, predisposing affected individuals to greater bacterial colonization and recurrent infection. High rates of dermatophytosis are seen as well.
2.4.2 Histology
Epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma) is characterized by orthokeratotic hyperkeratosis overlying the epidermis with marked alterations in the upper layers. The keratinocytes have marked clumping of keratohyalin granules and ballooning vacuolization. There is slight epidermal acanthosis (Figs. 2.11 and 2.12). There is continuous involvement of the epidermis unlike the situation in some epidermal nevi, in which focal epidermolytic hyperkeratosis occurs as an incidental finding [62]. A mild dermal inflammatory reaction is seen in some patients. Due to its striking and characteristic histologic findings, this is one type of ichthyosis that is readily diagnosed based upon routine microscopic examination.
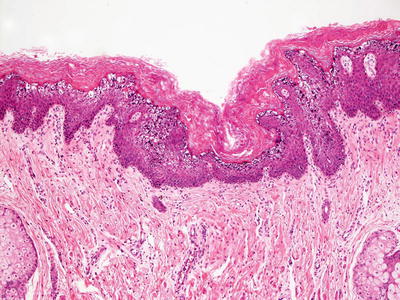

Fig. 2.11
Epidermolytic hyperkeratosis features marked clumping of keratohyaline granules in the granular layer of the epidermis
Fig. 2.12
Epidermolytic hyperkeratosis demonstrates marked hyperkeratosis, keratohyaline clumping and vacuolization of the superficial layers of the epidermis
Ichthyosis bullosa of Siemens is quite similar to bullous congenital ichthyosiform erythroderma, but clinically lacks the erythroderma [62]. Histologically, mild epidermolytic hyperkeratosis is detected and there is minimal dermal inflammatory reaction [63, 64]. Altered tonofilaments are present as seen by electron microscopy [64].
2.4.3 Pathogenesis
Bullous epidermolytic hyperkeratosis has an autosomal dominant pattern of inheritance. The classic generalized form of epidermolytic hyperkeratosis is caused by mutations in keratin 1 and keratin 10 [65–68]. Keratins 1 and 10 are parts of the keratin intermediate filament [11, 69, 70]. Mutations in either keratins disrupt keratin filaments, causing skin fragility with impaired skin barrier function. Localized forms of epidermolytic hyperkeratosis are due to genetic mosaicism in the affected tissue [11, 71].
2.5 Non-bullous Epidermolytic Hyperkeratosis/Congenital Ichthyosiform Erythroderma
2.5.1 Clinical Features
Congenital ichthyosiform erythroderma is a rare autosomal recessive non-syndromic ichthyosis [1]. Babies may present with a collodion membrane with subsequent development of generalized redness and fine white scales once the membrane is shed. Frequency of blistering improves after childhood, although subtle erythema and fine scaling may persist over an affected individual’s lifetime [72]. The degree of palmoplantar keratoderma varies with some individuals with no acral hyperkeratosis.
2.5.2 Histology
Congenital ichthyosiform erythroderma, also known as ichthyosis congenita type 1 , demonstrates marked hyperkeratosis and focally prominent parakeratosis, in contrast to the lamellar variant [73] (Fig. 2.13). Psoriasiform epidermal hyperplasia is present (Fig. 2.14). A mild superficial perivascular lymphocytic infiltrate is seen in some cases.

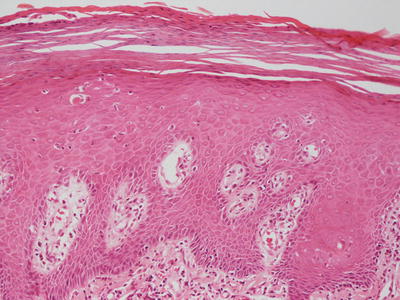
Fig. 2.13
Congenital ichthyosiform erythroderma is characterized by acanthosis with overlying confluent parakeratosis
Fig. 2.14
Congenital ichthyosiform erythroderma demonstrates confluent and thick parakeratosis overlying acanthotic epidermis with focal presence of a granular layer
2.5.3 Pathogenesis
The best characterized genetic defect in congenital ichthyosiform erythroderma is the transglutaminase-1 gene [47, 50]. Mutations in the ATP-binding cassette transporter ABCA12 have also been found in a small number of cases [61, 74, 75]. Other mutated genes implicated in this disorder are 12R-lipoxygenase (ALOX12B) and arachidonate lipoxygenase-3 (ALOXE3) [57]. These mutations which completely eliminate the enzyme function have been found in a small number of families in the Mediterranean and Germany [57, 76]. Functional studies have provided evidence that the lipoxygenases ALOX12B and ALOXE3 are important in skin differentiation, permeability barrier function, and the biological actions of essential fatty acids [77, 78].
2.6 Erythrokeratoderma Variabilis
2.6.1 Clinical Features
Erythrokeratoderma variabilis is an autosomal dominant disease that presents at birth or within the first year of life [22]. Patients have generalized fixed brown hyperkeratotic plaques with migratory erythematous, well-circumscribed patches and plaques that may be triggered by trauma or temperature variation. The hyperkeratotic plaques tend to persist and worsen during childhood, but stabilize later in life. Migratory lesions may resolve in adulthood following stabilization of the plaques [79].
2.6.2 Histology
Erythrokeratoderma variabilis has a nonspecific histologic pattern. Orthokeratotic hyperkeratosis is present with an acanthotic epidermis and prominent granular layer. Focal parakeratosis may be present. Follicular hyperkeratosis is described in some patients, while others demonstrate dyskeratosis [45, 80]. A mild lymphohistiocytic infiltrate permeates the superficial dermis in most cases. Immunohistochemistry shows a marked reduction in epidermal Langerhans cells [81]. Electron microscopic examination reveals membrane clefts resembling those seen in harlequin fetus [45].
2.6.3 Pathogenesis
Erythrokeratoderma variabilis is primarily an autosomal dominant disorder that is caused by de novo missense mutations in connexin 31 (encoded by GJB3 gene) [82]. Mutations in connexin 30.3 (encoded by GJB4 gene) and connexin 43 (encoded by GJA1) have also been reported in this disorder [83–85]. Connexins form important intercellular gap junctions that mediate the exchange of ions and metabolites between adjacent cells. They are important for the development and maintenance of the epidermis [86]. Mutations in connexins result in abnormal localization of the proteins and their aggregation within the Golgi, resulting in defects in gap junction formation [87].
2.7 Netherton Syndrome
2.7.1 Clinical Features
Netherton Syndrome is a rare autosomal recessive condition with no gender and ethnic predilection. It has a reported incidence of 1 in 200,000 newborns, accounting for approximately 18 % of all cases of congenital erythroderma [88]. Netherton Syndrome is characterized by ichthyosis, hair shaft abnormalities (specifically, trichorrhexis invaginata or “bamboo hairs ”) and atopy with elevated serum levels of immunoglobulin E [90–92]. Patients may present at birth with erythroderma or collodion membrane [22]. Ichthyosis varies from generalized fine scaling to plate-like lesions mimicking lamellar ichthyosis. Ichthyosis linearis circumflexa is pathognomonic in Netherton syndrome and is characterized by polycyclic, erythematous plaques with double-edged scale along the lesion edge. Some authors allow for the presence of congenital ichthyosiform erythroderma as a component of this syndrome as opposed to ichthyosis linearis circumflexa [89].
During the neonatal period, patients are at increased risk of hypernatremic dehydration, temperature dysregulation, failure to thrive, and infection . Patients also are at increased risk for systemic absorption of applied topical therapies [22]. Associated atopy may be more pronounced and persistent. Lastly, early onset of cutaneous neoplasms can be seen in some patients with Netherton syndrome, which demonstrate the same characteristics as similar neoplasms occurring in patients without the syndrome [93].
2.7.2 Histology
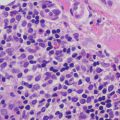
Ichthyosis linearis circumflexa shows focal parakeratosis alternating with orthokeratosis. Parakeratosis may be the dominant pattern in severe cases. The granular layer is markedly diminished or completely absent underlying the areas with parakeratosis, and slightly thickened in other regions [92]. Psoriasiform epidermal hyperplasia overlies a mild superficial perivascular lymphocytic infiltrate [94, 95] (Figs. 2.15, 2.16 and 2.17). The presence of neutrophils, even forming abscesses in some cases, results is a microscopic appearance quite similar to that seen in psoriasis [96]. However, suprapapillary thinning is not seen in most cases [97]. Round lipid bodies are seen within the parakeratotic cells in electron microscopy [98].
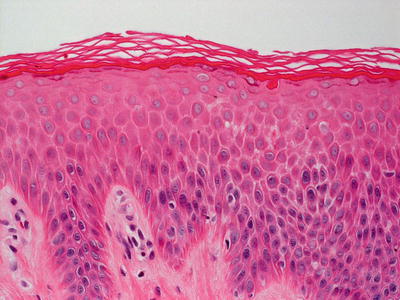


Fig. 2.15
Skin biopsies from patients with Netherton’s syndrome demonstrate ichthyosis linearis circumflexa with the characteristic psoriasiform and mildly spongiotic epidermis
Fig. 2.16
Ichthyosis linearis circumflexa demonstrates psoriasiform epidermal hyperplasia with overlying hyperkeratotic orthokeratosis and a variable dermal inflammatory infiltrate
Fig. 2.17
Psoriasiform epidermal hyperplasia with focal preservation of the granular layer and orthokeratotic hyperkeratosis is seen in ichthyosis linearis circumflexa
A subset of patients with Netherton syndrome has other histologic changes in skin biopsies. Prominent dermal eosinophils are seen in up to one third of patients with Netherton syndrome. Rarely, the histologic pattern is one of compact orthohyperkeratosis and a thickened granular layer [97].
2.7.3 Pathogenesis
Ichthyosis linearis circumflexa is inherited as an autosomal recessive trait. The disorder can present in isolation or, more commonly, as part of Netherton syndrome , an autosomal recessive disorder caused by mutations in the SPINK5 (serine protease inhibitor Kazal-type 5) gene that encodes for LEKTI (lymphoepithelial-Kazal-type 5 inhibitor) , a serine protease inhibitor [11, 88, 99]. Several cases of ichthyosis linearis circumflexa that carry SPINK5 mutations have been reported, suggesting that isolated ichthyosis can be a part of the Netherton syndrome phenotypic spectrum [100, 101].
LEKTI is normally expressed in the epidermis, within the stratum corneum and stratum granulosum [102, 103]. Mutations in LEKTI can result in unopposed serine protease activity, leading to desmosome degradation and premature desquamation of the epidermis. LEKTI functions by inhibiting serine proteases via regulation of activities of kallikrein 5 and kallikrein 7 in the epidermis, both of which degrade desmosomes in the skin [104–106]. Mutations in SPINK5 gene result in a truncated, nonfunctional LEKTI protein. Due to loss of functional LEKTI, there is increased activities of kallikrein 5, kallikrein 7 and epidermal elastase 2, resulting in loss of stratum corneum skin barrier function [99, 107, 108]. Some studies have provided evidence that elastase 2 is an epidermal protease important in the pathogenesis of Netherton syndrome [107]. Elastase 2 is constitutively expressed and active in the stratum corneum and stratum granulosum. It is involved in the processing of filaggrin and the formation of lipid lamellae , thus affecting skin barrier function.
In addition to inhibiting kallikreins , LEKTI has diverse inhibitory activities towards a number of other proteases, including trypsin, plasmin, subtilisin A, cathepsin G, and human neutrophil elastase [109, 110]. Taken together, it appears that the balance between proteases and protease inhibitors, such as LEKTI , is essential for normal skin desquamation and skin barrier function [111].
Different patients with Netherton syndrome can have different truncation mutations in SPINK5 [88, 111]. There might be a correlation between genotype and phenotype in Netherton syndrome, depending on the location of the mutations. It has been shown that the severity of the skin phenotype in this disorder correlates inversely with the levels of functional LEKTI protein and its serine protease inhibitory activity [112].
2.8 Refsum Disease
2.8.1 Clinical Features
Refsum disease is an uncommon, autosomal recessive disorder of lipid metabolism, typically affecting Scandinavians and Northern European populations [22]. Extracutaneous features include retinitis pigmentosa, polyneuropathy, cranial nerve disease, and cerebellar symptoms [22]. Following onset of neurologic and ophthalmologic disease, patients typically present with fine scales along the lower trunk and extremities that may progress to thicker, plate-like scaling if left untreated [113]. Clinical improvement may be achieved by eliminating chlorophyll, phytol, phytanic acid and their precursors from the diet.
2.8.2 Histology
Refsum syndrome demonstrates skin with orthokeratotic hyperkeratosis and acanthosis. Additionally, vacuolization is present within keratinocytes [114]. However, despite these histologic features, the diagnosis is more readily established with plasma phytanic acid studies than by skin biopsy [26, 115].
2.8.3 Pathogenesis
Refsum disease is a biochemical disorder caused by deficiency in the breakdown of phytanic acid. This is due to mutations in PHYH (peroxisomal enzyme phytanoyl-CoA hydroxylase) gene or PEX7 (peroxisomal biogenesis factor 7) gene that are involved in various aspects of phytanic acid metabolism [116–118].
In healthy individuals, phytanic acid undergoes oxidative decarboxylation by alpha-oxidation in the peroxisome to form formyl-CoA and pristanic acid. In Refsum disease, the alpha-oxidation of phytanic acid is impaired by mutations in either the PHYH gene that catalyzes this process or in PEX7 that is required for the correctly targeting of PHYH to the peroxisome [116–118]. As a consequence, phytanic acid fails to be properly metabolized and accumulates in the blood and tissues of the patients. High levels of phytanic acid can interfere with protein prenylation and impair the correct targeting of prenylated proteins to cellular membranes [119]. Phytanic acid is also a ligand for several members of the nuclear hormone family, including the retinoid X receptors (RXRs) and the peroxisome proliferator-activated receptors (PPARs) , which are involved in the regulation of lipid and glucose metabolism. Thus, defects in phytanic acid metabolism can result in a wide range of phenotypic abnormalities.
Infantile Refsum disease is distinct from classical Refsum disease. It is an autosomal recessive disorder of peroxisome biogenesis [120–122]. Biochemical derangements in this condition include increased levels of phytanic acid, pipecolic acid and very-long-chain fatty acids, as well as abnormalities in the synthesis of plasmalogen lipids [122]. Two of the genes implicated in infantile Refsum disease are PEX1 (peroxisomal biogenesis factor 1) and PEX6 (peroxisomal biogenesis factor 6) , which are ATPases and function in the import of peroxisomal proteins [123].
2.9 Sjogren–Larsson Syndrome
2.9.1 Clinical Features
Sjogren–Larsson syndrome is a rare disorder, characterized by progressive neurologic impairment with spastic paralysis and mental retardation in the setting of generalized fine, fish-like scaling and refractory pruritus [22]. Newborns may present with collodion membrane. Glistening white dots in the retina is pathognomonic of the disorder [22]. The disease course is progressive with most morbidity and mortality secondary to the associated neurologic disease.
2.9.2 Histology
In Sjogren–Larsson syndrome, the epidermis shows pronounced hyperkeratosis with only rare parakeratotic foci (Fig. 2.18). It is acanthotic and the granular layer is increased in thickness [124]. A papillomatosis growth pattern has been described [125]. There is a modest inflammatory infiltrate in the superficial dermis. Lamellar inclusions and increased numbers of mitochondria are seen in keratinocytes on electron microscopy [124, 126].
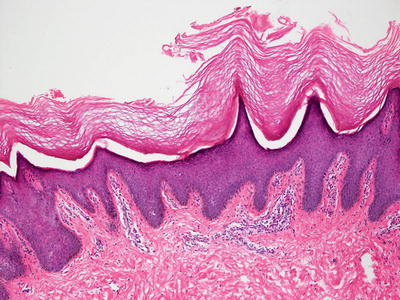
Fig. 2.18
Histologic findings include orthokeratotic hyperkeratosis, focal papillomatosis, and acanthosis in patients with Sjogren–Larsson syndrome
2.9.3 Pathogenesis
Sjogren–Larsson syndrome is an autosomal recessive disorder of lipid metabolism, caused by mutations in ALDH3A2 , a gene that encodes for the microsomal enzyme fatty aldehyde dehydrogenase (FALDH) , resulting in a deficiency of FALDH [127]. Over 70 mutations in ALDH3A2 gene have been identified in Sjogren–Larsson syndrome patients including amino acid substitutions, deletions, insertions, and splicing errors [128]. The function of FALDH is to catalyze NAD-dependent oxidation of medium- and long-chain fatty aldehydes to fatty acids [129, 130]. The pathogenesis of Sjogren–Larsson syndrome is a result of abnormal lipid accumulation in the membranes of skin and brain cells, the formation of aldehyde Schiff base adducts, and impaired eicosanoid metabolism [128]. Defects in the metabolism of leukotriene B4 , which is a pro-inflammatory mediator, also appears to play an important role in the disease [131].
Ultrastructural analysis of Sjogren–Larsson syndrome skin shows misshapen lamellar bodies and abnormal lipid inclusions in the cytoplasm of cells in the granular layer in the epidermis, indicating that affected keratinocyte membranes have an altered lipid composition as well as abnormal water barrier function that can cause reactive hyperkeratosis in the epidermis [124, 128, 132].
2.10 Conradi–Hunermann–Happle Syndrome/X-Linked Dominant Chondrodysplasia Punctata Type 2
2.10.1 Clinical Features
Conradi–Hunermann–Happle Syndrome is the X-linked dominant subtype of chondrodysplasia punctata, a varied mix of skeletal dysplasias arising from deficiencies in cholesterol biosynthesis. It is a rare genodermatosis, primarily affecting females [133]. Erythroderma may present soon after birth, followed by development of hyperkeratotic blaschkoid plaques [133]. These plaques evolve to form linear and whorled, atrophic and hypopigmented patches. Scalp alopecia is a frequent clinical feature as well. Additional clinical findings include cataracts, short stature, rhizomelic limb shortening, and vertebral anomalies with stippled epiphyses on X-ray. Overall, skin changes may improve with persistence of blaschkoid follicular atrophoderma and scarring alopecia.
2.10.2 Histology
Conradi–Hunermann–Happle syndrome associated with chondrodysplasia punctata demonstrates skin with thick laminated orthokeratosis. Prominent keratotic follicular plugs are present and give rise to dystrophic calcification, a finding that appears to be specific for this syndrome [134].
2.10.3 Pathogenesis
This syndrome is an X-linked dominant disease caused by mutations in emopamil binding protein (EBP , also called 3-beta-hydroxysteroid-delta8, delta7-isomerase ) located on the X chromosome [135, 136]. To date, over 70 different mutations in EBP have been reported.
EBP has dual functions. It serves as a binding protein for the calcium blocker emopamil, and also functions as a delta8–delta7 sterol isomerase [137, 138]. Defects in EBP result in increased levels of the sterol precursors 8(9)-cholesterol and 8-dehydrocholesterol. Measuring plasma sterol profiles is a useful test in the diagnostic work-up for Conradi–Hunermann–Happle syndrome, and there is a relationship between sterol levels and the disease phenotype [135, 136, 139, 140].
2.11 Other Syndromes with Ichthyosis
CHILD (congenital hemidysplasia with ichthyosiform erythroderma and limb dysplasia) syndrome is an X-linked disorder with a remarkable feature of an inflammatory nevus with unique lateralization and midline demarcation. Histologic features of the nevus show alternating areas of orthokeratotic hyperkeratosis and parakeratosis. In the areas with parakeratosis, ghost cells and other keratinocytes demonstrating large granular cells are seen. Vacuoles are present within the cytoplasm of these cells. Ultrastructural analysis reveals many cytoplasmic lamellar bodies [141]. Similar structures are seen in an extracellular location [142]. Verruciform xanthoma has been described in association with this type of ichthyosis [143–145].
IBIDS syndrome is a syndrome with ichthyosis, brittle hair (trichothiodystrophy), impaired intelligence, decreased fertility and short stature [146]. The ichthyosis is the lamellar type and can range from mild to severe.
MAUIE syndrome is comprised of micropinnae, alopecia universalis, ichthyosis and ectropion [147]. The histologic findings include orthokeratosis alternating with parakeratosis. The granular layer is diminished or absent and the epidermis is acanthotic. Focal vacuolization of superficial keratinocytes may be present. Dilated capillaries are surrounded by a mild perivascular lymphohistiocytic inflammatory infiltrate. Clinical areas of skin sparing demonstrate mild orthokeratosis but no other histologic changes. There is a suggestion that patients with MAUIE syndrome are predisposed to developing skin cancers at a young age, although there is no good explanation for this observation [147].
2.12 Seborrheic Dermatitis
2.12.1 Clinical Features
Seborrheic dermatitis is a multifactorial disorder affecting cutaneous surfaces with the highest density of sebaceous glands, namely the scalp, face (nasolabial folds, ears, eyebrows, and glabella), upper chest/presternal area, and intertriginous areas. It is seen more commonly in males, and across various ethnic and racial backgrounds [148]. Current opinion suggests that excessive colonization by malassezia species, a lipophilic yeast, may play a significant role in the development and chronicity of the disease. It affects 1–3 % of immunocompetent adults, but prevalence rates are as high as 83 % in persons with immunodeficiency, implicating immune dysregulation as a potential pathogenic factor [148]. In children, there is a bimodal distribution with incidence highest in infants less than 3 months old and in adolescents, suggesting an androgenic hormonal influence as well.
On physical exam, patients present with erythematous plaques and patches with overlying greasy, yellow scale in any of the above mentioned sebaceous gland-rich body areas (Fig. 2.19). Additional clinical findings may include erythematous folliculitis. Pruritus is typically minimal, distinguishing it from atopic dermatitis. The disorder is amenable to a number of topical therapies, but it has a chronic and relapsing course.
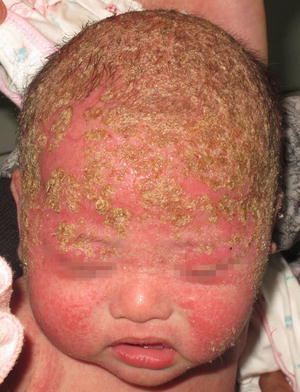
Fig. 2.19
Seborrheic dermatitis presents as rough, erythematous thin plaques with overlying adherent, greasy scales involving the scalp and face of an infant (photo courtesy of Minh Hoang, MD, Ho Chi Minh City, Vietnam)
2.12.2 Histology
Seborrheic dermatitis has parakeratosis that is often most prominent in the outflow tracts of hair follicles (Fig. 2.20). Small clusters of neutrophils may be present in this location (Fig. 2.21). The epidermis demonstrates psoriasiform hyperplasia with bulbous elongation of rete ridges. There is focal hypogranulosis in most cases [149]. Some intraepidermal spongiosis is present, especially in regions surrounding and within the follicular epithelium (Fig. 2.22). Classically, this is most pronounced in regions of the follicular ostia and/or infundibula. Papillary dermal edema may be observed in conjunction with vascular dilation. The dermis is characterized by an infiltrate of lymphocytes, histiocytes and rare neutrophils concentrated around pilosebaceous units. Commonly, colonization with Pityrosporum (Malassezia) yeast forms is present, especially within the follicular keratin and stratum corneum.
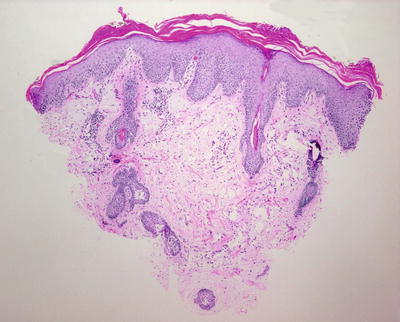
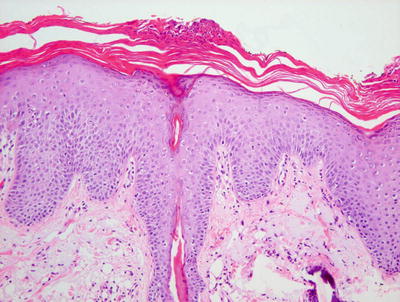
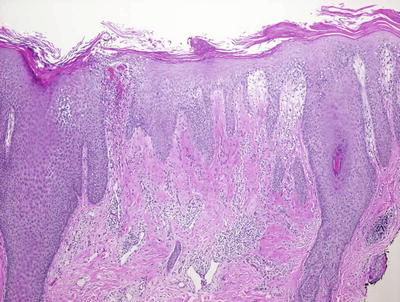
Fig. 2.20
Seborrheic dermatitis demonstrates tufts of parakeratosis at the outflow tracts of hair follicles and psoriasiform epidermal hyperplasia
Fig. 2.21
Clusters of neutrophils are seen overlying follicular orifices in some cases of seborrheic dermatitis
Fig. 2.22
Spongiosis, especially within follicular epithelium, and psoriasiform epidermal hyperplasia are seen along with a mild lymphocytic infiltrate in seborrheic dermatitis
It is often quite difficult to distinguish seborrheic dermatitis from psoriasis. The perifollicular nature of the infiltrate, small amounts of spongiosis and partial retention of the granular layer are features in favor of seborrheic dermatitis , although these can all be seen in some cases of psoriasis as well.
2.12.3 Pathogenesis
Pathologic features in seborrheic dermatitis include androgenic hormones, fungal colonization and immunologic regulation. In fact, there is a strong correlation between yeast density on the skin and the severity of seborrheic dermatitis, a concept supported by the fact that antifungal agents are routinely used effectively in the treatment of seborrheic dermatitis [150, 151]. A reaction between Malassezia , cells in the stratum corneum where the yeasts reside, and the immune system may be a primary trigger factor [152]. Moreover, the presence of host susceptibility factors associated with Malassezia infection and the host immune response may predispose certain individuals to developing seborrheic dermatitis. How Malassezia yeasts initiate inflammation in seborrheic dermatitis is not clear. The yeasts may cause inflammation by inducing cytokine production by keratinocytes or Langerhans cells with subsequent T-lymphocyte activation [153]. Another factor that may cause inflammation is the lipase activity of Pityrosporum ovale, which generates inflammatory fatty acids from skin lipids [154].
Scalp hyperkeratosis in seborrheic dermatitis , commonly known as dandruff , is believed to be mediated by Malassezia metabolites, particularly irritating free fatty acids released from sebaceous glands [155]. Transcriptome microarray analysis of dandruff lesional scalp skin as compared with non-dandruff scalp skin shows that genes involved in lipid metabolism are strongly reduced, and genes involved in the immune response defense against pathogens are increased in dandruff lesional skin as compared with normal skin [156]. Interestingly, treatment of scalp dandruff with a shampoo containing potentiated zinc pyrithione (ZPT) shows that the treatment induces a transcriptomic profile resembling that of normal scalp skin .
2.13 Psoriasis
2.13.1 Clinical Features
Psoriasis is a systemic inflammatory condition that is seen across the pediatric spectrum from infancy to adolescence. It is thought to represent a Type 1 T-helper cell predominant hyperactivity in response to environmental stimuli in genetically susceptible individuals. Psoriasis is estimated to affect 1 % of the general population with a mean age of onset between 8 and 11 years old when seen in children [157]. A family history of psoriasis appears to convey a significant risk for development of the disease in children with 68 % of children having an affected family member in one study [158].
Erythematous plaques with overlying micaceous scale at the extensor extremities and scalp characterize chronic plaque type psoriasis, although any cutaneous surface can be involved (Fig. 2.23). Plaque type psoriasis is the most common subtype seen in children [157]. The scalp is the most frequently affected area, and it is the site of initial presentation in more than 50 % of pediatric patients [159]. Guttate psoriasis is seen in children as well with rates as high as 40 %. In this variant, the physical exam is noted for smaller erythematous, scaled papules and plaques in a generalized distribution, including the face. Skin changes often follow Group A β-hemolytic streptococcal infection . In infants, psoriatic diaper dermatitis (napkin dermatitis ) may occur with circumscribed, erythematous plaques at the lower abdomen, groin, and buttocks. Other subtypes include intertriginous psoriasis (minimally scaled, erythematous patches and plaques at the flexural folds) , pustular psoriasis (superficial sterile pustules overlying erythematous patches and plaques possibly with accompanying fever, arthralgia), palmoplantar psoriasis , anogenital psoriasis (eroded patches or hyperkeratotic plaques at the groin and perianal skin), erythrodermic psoriasis (generalized erythema with secondary desquamation) (Fig. 2.24), and even isolated nail psoriasis.
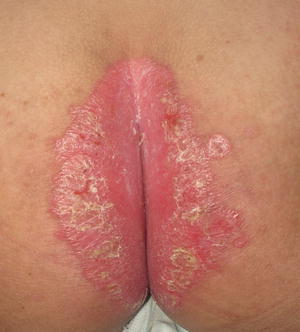
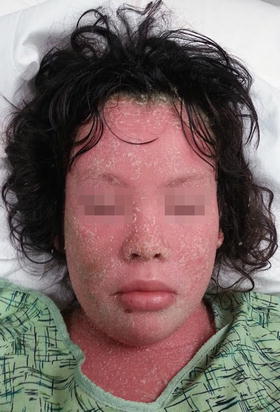
Fig. 2.23
Psoriasis presents as a well demarcated erythematous plaque with micaceous scale along the gluteal fold (photo courtesy of Minh Hoang, MD, Ho Chi Minh City, Vietnam)
Fig. 2.24
Erythrodermic psoriasis may present as generalized erythema with desquamation as seen in this teenage girl
For most patients, psoriasis is a chronic disease with periodic episodes of remission and relapse [159]. The relationship between the cutaneous disease and systemic inflammation has long been evidenced by the presence of psoriatic arthritis. Data now also link psoriasis with metabolic syndrome and cardiovascular disease with increased incidence of hyperlipidemia, obesity, hypertension, and diabetes mellitus even in affected children [157].
2.13.2 Histology
The histologic features of psoriasis are manifold depending upon the type and the duration of the lesions biopsied [160]. Classic plaque psoriasis is characterized by confluent parakeratosis that contains clusters of neutrophils (Munro’s microabscesses ) (Figs. 2.25 and 2.26). The underlying epidermis displays marked hypogranulosis and regular acanthosis with “psoriasiform” elongation of the rete ridges. Neutrophilic clusters (spongioform pustules of Kogoj) are also present within the epidermis. The most prominent dermal changes include suprapapillary thinning with marked dilatation of post-capillary venules. A modest superficial perivascular infiltrate of lymphocytes and histiocytes is present, and eosinophils are uncommon in psoriasis [161]. Sebaceous gland atrophy has been described in a series of patients with psoriasis involving the scalp [162].
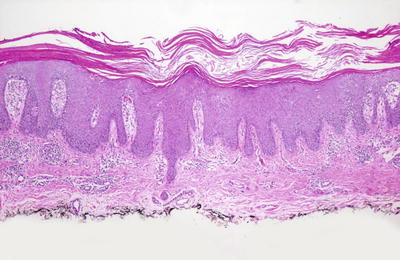
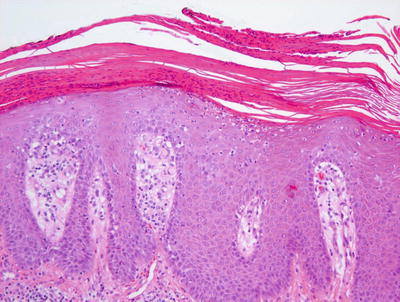
Fig. 2.25
Thick parakeratosis overlies an acanthotic epidermis with psoriasiform epidermal hyperplasia in psoriasis
Fig. 2.26
Neutrophilic clusters within the stratum corneum and in the uppermost epidermis that lacks the granular layer characterize psoriasis
Large neutrophilic abscesses within the stratum corneum as well as within the epidermis characterize pustular psoriasis. The other histologic changes as described above may or may not be present since these lesions are often quite acute. In contrast to plaque lesions, focal spongiosis may be present, acanthosis is rarely pronounced and elongation of the rete ridges is not usually regular or prominent. The dermal infiltrate may include neutrophils. Vascular dilation is usually present in these cases [163].
Guttate psoriasis demonstrates yet another constellation of histologic findings. Parakeratosis is present, yet it is often focal and relatively sparse. It may be difficult to find neutrophils in the stratum corneum and they are most often not seen within the epidermis. The epidermis is only minimally acanthotic and elongation of rete ridges is not normally present (Fig. 2.27). Vascular dilation is invariably present and is often a significant clue to establishing this diagnosis (Fig. 2.28).
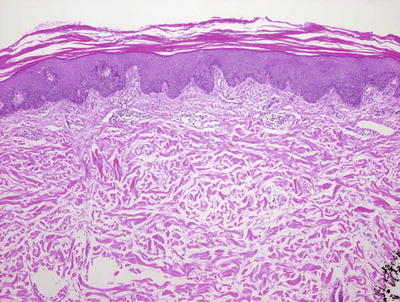
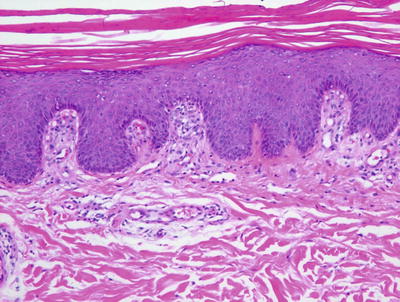
Fig. 2.27
Guttate psoriasis demonstrates an acanthotic epidermis with focal parakeratosis. The rete ridges are not as elongates as they are seen in plaque psoriasis
Fig. 2.28
Focal parakeratosis, focal preservation of the granular layer and ectatic blood vessels within papillary dermal tips characterize guttate psoriasis
The differential diagnosis depends upon the type of psoriatic lesion biopsied. Classic plaque type psoriasis can be difficult to distinguish from a chronic spongiotic dermatitis, though the presence of confluent parakeratosis, neutrophils and vascular dilation and the absence of a granular layer and spongiosis may help one to make a diagnosis of psoriasis. If lesions of psoriasis are secondarily traumatized, this distinction can be very difficult, if not impossible. Pityriasis rubra pilaris shares some histologic features with plaque psoriasis. However, the parakeratosis is less confluent and is restricted to the outflow tracts of hair follicles in most cases. The epidermis is less acanthotic with less elongation of the rete ridges. The granular layer is preserved in much of the epidermis and the inflammatory infiltrate within the dermis tends to be less pronounced [164].
Pustular psoriasis closely resembles the histologic changes seen in acute generalized exanthematous pustulosis . The presence of eosinophils and spongiosis would favor acute generalized exanthematous pustulosis, but the distinction can be difficult without clinical history (Fig. 2.29). Subcorneal pustular dermatosis may demonstrate similar if not identical features to that of psoriasis . In some cases, IgA pemphigus can display clusters of intraepidermal neutrophils, but the presence of focal acantholysis will help with this distinction. Also in the differential diagnosis is dyshidrotic eczema, although it is usually characterized by a greater degree of spongiosis [163].
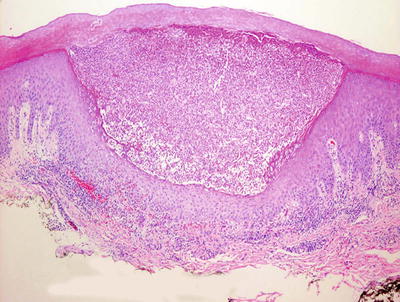
Fig. 2.29
A subcorneal neutrophilic abscess in pustular psoriasis . Other changes associated with psoriasis may not be present in this acute process
Guttate psoriasis raises the differential diagnoses of pityriasis rosea and small plaque parapsoriasis (guttate parapsoriasis, digitate dermatosis). The presence of neutrophils and focal loss of the granular layer favor psoriasis. The presence of tufts of parakeratosis and focal hemorrhage within the epidermis and dermis favors pityriasis rosea. The changes in small plaque parapsoriasis are less intense than those seen in psoriasis, and there may be slightly more exocytosis of lymphocytes into the epidermis; however, these changes can be very subtle and it may be difficult to distinguish this entity from psoriasis without clinical history and follow-up.
2.13.3 Pathogenesis
Psoriasis is a complex disease whose pathogenesis is multifactorial with genetic, microbial and immunologic components. Studies of the genetic basis of psoriasis have shown that the psoriasis susceptibility locus 2 (PSORS2) is located on chromosomal region 17q25-qter in a large family of European ancestry [165]. Sequencing of the locus revealed a mutation in CARD14 (caspase recruitment domain family, member 14) that segregates with psoriasis [166]. As shown in the study, psoriasis skin has reduced levels of CARD14 in the basal layer, but increased levels in the upper layers of the epidermis as compared with normal skin [166]. It has been suggested that mutations in CARD14 can upregulate an inflammatory response via the nuclear factor-κB (NF-κB) pathway.
Homozygous missense mutation in IL36RN (interleukin-36 receptor antagonist) gene has been identified in generalized pustular psoriasis [167, 168]. IL36RN encodes for an anti-inflammatory cytokine, and substitution of a proline residue for leucine (L27P) has been predicted to affect the stability of IL-36RN and its interaction with its receptor (IL-1 receptor–related protein 2). The result of the mutation is reduced anti-inflammatory response and enhanced production of pro-inflammatory cytokines in the IL-1 family [167, 169].
Psoriasis is strongly associated with streptococcal infection, and patients have increased occurrence of the disease after such infections. It has been suggested that streptococcal association in psoriasis may reflect the co-occurrence of streptococcal determinants and superantigen production promoting skin homing of T lymphocytes in the immunopathogenesis of psoriasis [170].
The immunological basis of psoriasis involves mainly type 1 T-helper cells (Th1) and type 17 T-helper cells (Th17). Macrophages and dendritic cells produce interleukin-12 (IL-12) , which is important in inducing the differentiation of naïve CD4+ T lymphocytes into Th1 cells [171, 172]. Th1 cells produce Th1-type cytokines, including interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), and interleukin-2 (IL-2), in psoriatic skin lesions [173–175]. Serum levels of these cytokines have been shown to correlate with the severity of psoriasis [173, 176]. IFN-γ is important in the development of Th1-mediated immune response. TNF-α affects the proliferation and activation of keratinocytes and T lymphocytes in psoriasis [174, 177].
Th17 cells play a crucial role in T cell-mediated adaptive immunity in psoriasis [178, 179]. Naïve CD4+ T lymphocytes are stimulated to differentiate into activated Th17 cells by IL-1, IL-6, and transforming growth factor-beta (TGF-beta) . Th17 cells in turn produce IL-17 and IL-22, which are key cytokines that affect keratinocyte differentiation and induce chemokine expression in keratinocytes [180–182]. IL-17 and IL-22 contribute to the inflammatory process in psoriasis by inducing the accumulation of T lymphocytes and neutrophils in psoriatic lesions [183–185]. Dendritic cells and macrophages produce a key cytokine IL-23 that induces the proliferation and activation of Th17 cells [186]. High levels of IL-23 also stimulate the production of TNF-α and IL-22, which can induce the onset of psoriasis [174, 187]. Thus, the IL-23–Th17 axis appears to be crucial in the pathogenesis of psoriasis.
A number of other factors have been implicated in the pathogenesis of psoriasis. Studies have indicated a causal role of TGF-β in psoriasis [188]. TGF-β inhibitors (such as Etanercept ) have been used to treat psoriasis and other autoimmune diseases. NF-κB is another crucial mediator in psoriasis by modulating inflammation [189]. NF-κB may be involved in altered keratinocyte and immune cell behavior seen in psoriasis. Proangiogenic factors, namely angiopoietin and vascular endothelial growth factor (VEGF) , are involved in the induction of epidermal hyperplasia and angiogenesis in psoriasis [190, 191].
2.14 Reactive Arthritis (Reiter’s Disease)
2.14.1 Clinical Features
Reactive arthritis is defined by the triad of conjunctivitis, urethritis and arthritis. It most frequently occurs in young males within 4 weeks of an infection of the urogenital or gastrointestinal system. Approximately half of patients with reactive arthritis have mucocutaneous symptoms, most commonly circinate balanitis of the penis (50 %) and keratoderma blennorrhagicum in the skin (10 %) [195]. Findings of nail dystrophy (pustules along the nail folds, subungual debris, nail plate thickening, nail pitting, onycholysis, and leukonychia), ulcerative vulvitis (similar in appearance to circinate balanitis in males) and painless oral mucosal erosions have been reported as well [195]. Additional systemic manifestations include fever, fatigue, weight loss and visceral disease.
Keratoderma blennorrhagicum typically begins as erythematous, vesiculopustules with eventual evolution to hyperkeratotic plaques on the palms and soles [195]. These lesions also can be seen on the scalp, trunk and dorsal aspect of the extremities, mimicking pustular psoriasis . A similar process occurs on the penis (circinate balanitis) with uncircumcised males developing small fragile vesicles and pustules that evolve into painless, serpiginous erosions. Circumscribed males, in contrast, more commonly develop thick plaques on the glans penis. The clinical course is variable, either resolving spontaneously or progressing to a more chronic variant with intermittent flares. Reports suggest that children tend to have milder disease with fewer episodes of recurrence [195].
2.14.2 Histology
The histologic changes seen in Reiter’s disease are indistinguishable from those seen in pustular psoriasis [196]. Subcorneal and intraepidermal neutrophilic abscesses are seen in great abundance. Confluent parakeratosis overlies a variably acanthotic epidermis that has psoriasiform hyperplasia. Suprapapillary thinning and vascular ectasia are present along with a dermal infiltrate of lymphocytes, histiocytes and neutrophils [197, 198].
2.14.3 Pathogenesis
Reactive arthritis is thought to be secondary to an autoimmune immunologic response to an infectious insult in genetically predisposed individuals [195]. There is a strong association between disease presence and HLA-B27 haplotype , which is a MHC class I molecule involved in T-cell antigen presentation [199, 200]. On average, about 70–80 % of patients with the disease carry HLA-B27 [201]. In a study of Tunisian patients, other HLA alleles, namely HLA-B51 and HLA-DRB1, also were found to be associated with reactive arthritis [202].
Pathogens such as group A streptococci, Shigella, Salmonella, Yersinia, and Campylobacter as well as Chlamydia trachomatis have been implicated as triggering agents in reactive arthritis [203–205]. Group A streptococcal infection with elevated anti-streptolysin O antibodies can cause post-streptococcal reactive arthritis [204]. How the interaction of the inciting microorganism with an HLA-B27 -positive host leads to disease development is not known. It has been suggested that molecular similarities (molecular mimicry) between the inciting microorganism antigen and HLA-B27 molecule may trigger an immune response that results in reactive arthritis [199]. Microbial antigens may also utilize toll-like receptors (TLRs) of the innate immune system to incite the disease process [206, 207]. Synovitis in reactive arthritis is induced by proinflammatory cytokines , including interleukin-17 (IL-17), IL-6, IL-1β, and IL-21. IL-17, which is produced by type 17 T-helper cells, is elevated in the synovial fluid of patients with the disease [208, 209].
2.15 Pityriasis Rubra Pilaris
2.15.1 Clinical Features
Pityriasis rubra pilaris is seen infrequently in children. It is divided into four categories with types III-V listed as juvenile variants. There is no gender predilection, and most pediatric cases are sporadic [210]. Pityriasis rubra pilaris is characterized by the cephalocaudal spread of coalescing, hyperkeratotic, follicular, salmon–colored papules with the eventual development of “islands of sparing” and the potential for generalization to erythroderma (Figs. 2.30, 2.31, and 2.32). A waxy, orange-yellow colored palmoplantar keratoderma may be present as well (considered a hallmark of juvenile disease by some experts) with or without associated nail dystrophy [211]. Many reports suggest an excellent response to retinoid therapy, but recurrence rates may be 20 % [210].
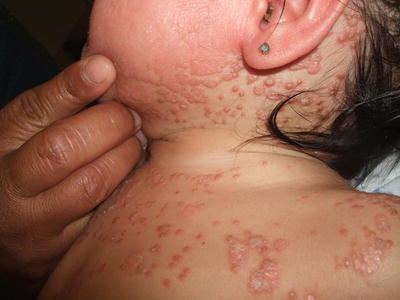
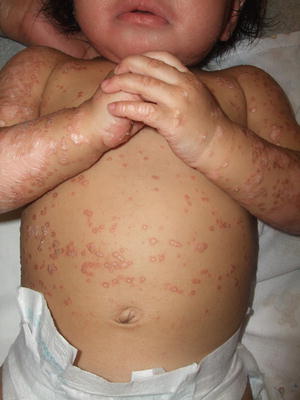
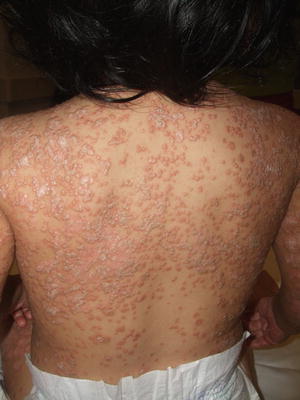
Fig. 2.30
Pityriasis rubra pilaris is characterized by coalescing hyperkeratotic, follicular, salmon-colored papules
Fig. 2.31
Pityriasis rubra pilaris may be widespread with cephalocaudal spread
Fig. 2.32
Islands of sparing are a key feature of pityriasis rubra pilaris
2.15.2 Histology
Pityriasis rubra pilaris is characterized by a distinct pattern of alternating parakeratosis and orthokeratosis. Some authors describe this pattern as occurring in a horizontal, as well as a vertical manner, giving rise to a “checkerboard” appearance. The areas of parakeratosis correlate with the follicular ostia, though this is not always seen on routine sectioning (Figs. 2.33, 2.34 and 2.35). The granular layer is present throughout, though focally diminished and in some areas may actually be of increased thickness. The epidermis is diffusely acanthotic [164], including in the areas of the suprapapillary plates and demonstrates psoriasiform epidermal hyperplasia. Focal acantholysis has been described in up to 70 % of lesions. The dermis shows a very sparse lymphohistiocytic inflammatory response. There is slight dilation of post-capillary venules in the papillary dermal tips. Hypertrophy of the arrector pili muscles is seen in some cases.
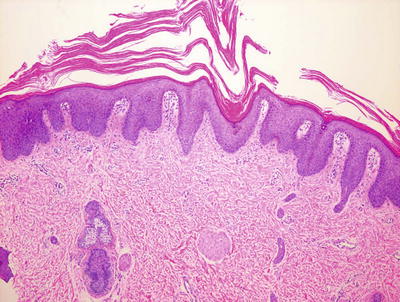
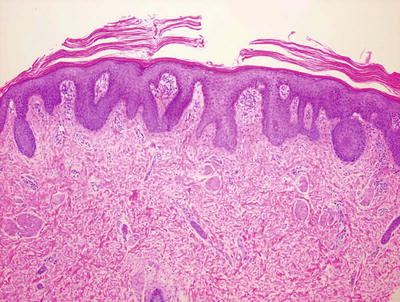
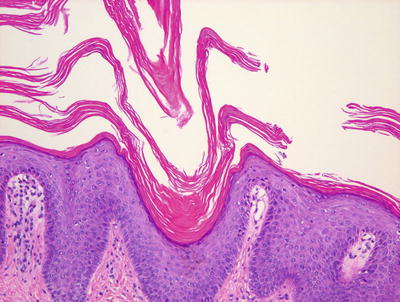
Fig. 2.33
Pityriasis rubra pilaris is characterized by psoriasiform epidermal hyperplasia, alternating parakeratosis and hyperkeratosis, and follicular dilatation
Fig. 2.34
Parakeratosis is not confluent in pityriasis rubra pilaris and psoriasiform epidermal hyperplasia coexists with a focally persistent granular layer
Fig. 2.35
Pityriasis rubra pilaris demonstrates parakeratosis at the outflow tract of dilated hair follicles
The main histologic differential diagnosis is psoriasis. Psoriasis differs by demonstrating confluent parakeratosis, neutrophils within the stratum corneum and epidermis, and more prominent elongation of rete ridges with suprapapillary thinning. Psoriasis also has a more intense lymphohistiocytic infiltrate in the dermis, often admixed with neutrophils .
Mycosis fungoides may also enter into the histologic differential diagnosis. This epidermotrophic lymphoma shows more invasion of the epidermis by atypical, enlarged and hyperchromatic lymphocytes than would be expected in pityriasis rubra pilaris [212]. In addition, the patterned parakeratosis is not a feature seen in mycosis fungoides .
2.15.3 Pathogenesis
Pityriasis rubra pilaris is often sporadic, but rare familial cases with autosomal dominant inheritance have been reported [213, 214]. Familial pityriasis rubra pilaris is caused by gain-of function mutations in CARD14 (caspase recruitment domain family, member 14) gene with increased expression of CARD14 in the epidermis of patients [215, 216]. CARD14 is an activator of nuclear factor (NF)-κB signaling , leading to the activation of the inflammatory response [217]. CARD14 does not appear to be involved in sporadic cases, as no mutations in CARD14 were found in a study of 61 patients with sporadic disease [218]. Another postulation of the etiology of pityriasis rubra pilaris is an abnormal immune response to an antigenic trigger. There are case reports of the disease occurring after streptococcal infections in children [219].
2.16 Pityriasis Lichenoides
2.16.1 Clinical Features
Pityriasis lichenoides is a disorder of unknown etiology currently thought to be a reactive T-cell lymphoproliferative disorder secondary to an antigenic stimulus [220]. In pediatric populations, disease before 2 years of age is exceedingly rare with bimodal peak incidence around 5 and 10 years of age [220]. In a retrospective cohort study by Ersoy-Evans et al., [220] patients with pityriasis lichenoides et varioliformis acuta (PLEVA) were slightly younger than those with pityriasis lichenoides chronica (PLC) . Outbreaks occurred most often in the winter and fall, with one third of patients reporting a history of preceding infection, primarily viral. No gender, ethnic, or geographic predilection has been identified in adults [221], but a male predominance is seen in children [220].
PLEVA is characterized by the acute onset of papulovesicles with secondary necrosis, focal ulceration, and development of overlying hemorrhagic crust (Fig. 2.36). Lesions tend to heal with scarring and post-inflammatory dyschromia. Patients with PLEVA frequently have associated fever and arthralgias [220].
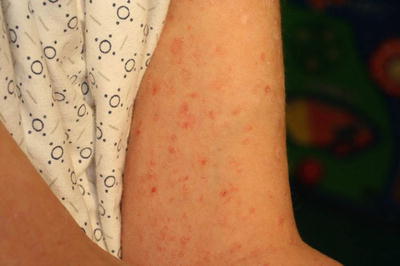
Fig. 2.36
Pityriasis lichenoides et varioliformis acuta (PLEVA) shows papulovesicles with secondary necrosis and overlying hemorrhagic crust
PLC has a more indolent course of flares and remission with recurrent crops of polymorphic scaly papules and thin plaques. Active lesions resolve with hypopigmentation (Fig. 2.37). In both disorders, most patients have generalized eruptions with involvement of the trunk and extremities [220].
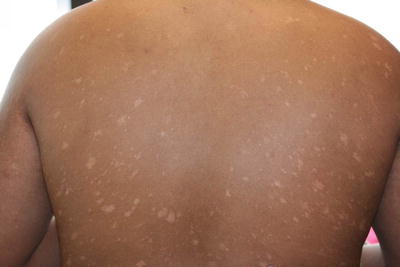
Fig. 2.37
Pityriasis lichenoides chronica (PLC) often presents as crops of polymorphic, hypopigmented papules and thin plaques
Overall, pediatric pityriasis lichenoides is recognized as a benign disorder with spontaneous resolution within years. However, as there are reports of malignant transformation to cutaneous T-cell lymphoma, long-term surveillance is recommended [221].
2.16.2 Histology
PLEVA is characterized by a thick parakeratotic stratum corneum (Fig. 2.38). The epidermis is variably atrophic and acanthotic, depending upon the developmental stage of the lesion sampled. Secondarily rubbed lesions may demonstrate psoriasiform epidermal hyperplasia. Dying keratinocytes are present within the epidermis, and marked basal vacuolization is present in florid cases (Figs. 2.39 and 2.40). In some cases, there is extensive epidermal necrosis that may result in ulceration. A superficial and occasionally deep infiltrate of lymphocytes is present with marked exocytosis into the epidermis and follicular epithelium . There is extravasation of erythrocytes within the papillary dermis, often extending into the epidermis (Fig. 2.41). Eosinophils are not frequently encountered.
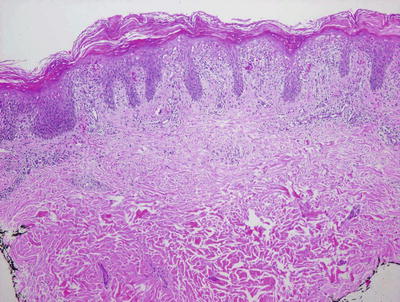
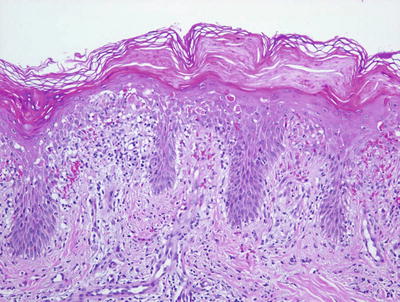

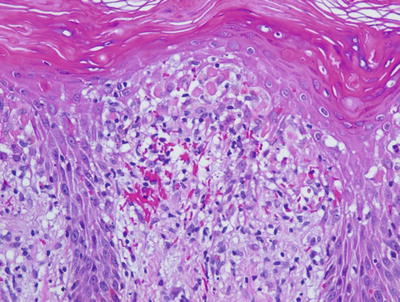
Fig. 2.38
Confluent parakeratosis overlies a brisk interface dermatitis in well-developed lesions of PLEVA
Fig. 2.39
PLEVA shows an intense interface dermatitis with abundant dying keratinocytes
Fig. 2.40
Dying keratinocytes and dermal hemorrhage are constant features in PLEVA
Fig. 2.41
Abundant dying keratinocytes may give rise to subepidermal blisters in PLEVA. Hemorrhage within the dermis and extending into the epidermis is almost always present
The histologic differential diagnosis of PLEVA includes pityriasis lichenoides chronica, pityriasis rosea, guttate psoriasis, digitate dermatosis (small plaque parapsoriasis), arthropod bite reaction, and lymphomatoid papulosis [222]. Pityriasis lichenoides chronica differs from PLEVA mainly in degree of inflammation. Similar histologic changes are present, but with much less intensity such that there is only focal parakeratosis, dying keratinocytes are rare, lymphocytes within the epidermis are sparse and hemorrhage is not often present within the dermis [223, 224]. Pityriasis rosea demonstrates only focal mounds of parakeratosis. In general, there is more prominent spongiosis (though only focally) in pityriasis rosea than is the case with PLEVA and dying keratinocytes are quite unusual. The dermal inflammatory infiltrate is less intense, though extravasated erythrocytes are commonly seen. Lesions of guttate psoriasis frequently demonstrate ectatic vessels located along the dermal epidermal junction, a characteristic feature that is helpful in making the distinction. Also, the presence of neutrophilic abscesses encountered in psoriasis, if present, will help in this differential diagnosis. The changes of digitate dermatosis are nonspecific and generally relatively mild. Delicate parakeratosis is present focally overlying small regions of mild spongiosis with a scant exocytosis of lymphocytes into the epidermis. The dermal infiltrate is superficial and of lesser intensity than that seen in PLEVA. Arthropod bite reactions may also have extensive histologic overlap with PLEVA. Parakeratosis is usually more focal in these lesions. Dermal hemorrhage is present in the region of the puncture wound and eosinophils are commonly encountered around dermal blood vessels as well as within the interstitial collagen. Lymphomatoid papulosis has virtually all of the features seen in PLEVA, and is differentiated mainly based upon the cytologic features of the lymphoid infiltrate . A minority population of dermal lymphocytes has cytologic atypia, resembling either the cells seen in Hodgkin’s disease or those seen in mycosis fungoides. CD30 immunostain may be helpful in highlighting these cells, and when they are present in clusters or in large numbers, this favors a diagnosis of lymphomatoid papulosis over that of PLEVA [225].
PLC has histologic changes that suggest a milder form of PLEVA [224]. Focal and delicate parakeratosis is present overlying an epidermis that contains minimal spongiosis and very rare dying keratinocytes [226] (Figs. 2.42 and 2.43). Scattered lymphocytes extend from the dermis into the epidermis. Basal vacuolization may be present but is relatively inconspicuous in most cases. Within the dermis, there is a mild lymphohistiocytic inflammatory infiltrate surrounding the superficial vascular plexus. In most cases, extravasated erythrocytes are minimally present or not seen. Eosinophils are not ordinarily encountered. The majority of the T cells are helper T cells. It has been suggested that the etiology for this eruption may be diverse, including reactions to viral infections [223, 227].
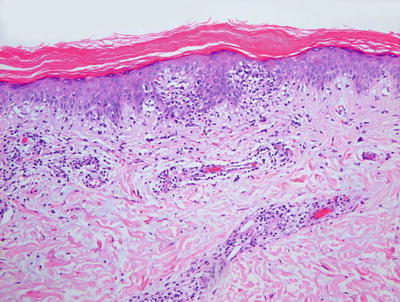
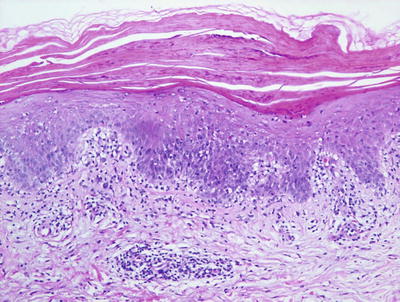
Fig. 2.42
PLC demonstrates parakeratosis, pityriasiform dermatitis, and slight amounts of dermal hemorrhage in some cases
Fig. 2.43
Exocytosis of lymphocytes into the epidermis with overlying parakeratosis is seen in PLC
The histologic differential diagnosis of PLC includes PLEVA, in which all of the same histologic changes are present much more extensively [224]. Also included is guttate psoriasis that has papillary dermal vascular ectasia and a more regularly acanthotic epidermis. Digitate dermatosis can be very difficult to distinguish from PLC and clinical correlation is essential to definitively separate these two entities. Pityriasis rosea demonstrates more spongiosis that is focal with mounds of parakeratosis limited to the areas of spongiosis and intraepidermal hemorrhage . Increased spongiosis is also seen in small plaque or guttate parapsoriasis [226].
2.16.3 Pathogenesis
PLEVA has been proposed to be part of a spectrum of clonal T-cell lymphoproliferative disorders. Studies of PLEVA patients have reported the presence of a dominant T-cell clone and monoclonal TCR-γ gene rearrangements [228–230]. CD8+ cytotoxic suppressor T lymphocytes are the main cell type in the inflammatory infiltrate in PLEVA unlike PLC, which has more CD4+ lymphocytes and FOXP3+ regulatory T-cells [223, 225, 231, 232]. Lymphoma has been reported in children following PLEVA eruption [233]. However, it remains controversial whether PLEVA has some malignant potential with possible progression to lymphoma.
Immune complex-mediated vasculitis has been proposed to be a possible etiology of pityriasis lichenoides. Immunofluorescence stains in biopsy specimens of fresh purpuric lesions obtained from patients with pityriasis lichenoides showed circulating C3 immune complex deposition in the basement membrane in some patients [234, 235].
Another proposed etiology of PLEVA is viral infection. Parvovirus B19 DNA was detected in some patients, and the incorporation of viral DNA into the host cell may result in keratinocyte antigenicity [236]. Another viral agent implicated in PLEVA is human herpesvirus-8 [223].
The inflammatory infiltrate in PLC is mainly composed of CD4+ T lymphocytes [237, 238]. Some studies have suggested that PLC is a form of indolent cutaneous T-cell dyscrasia with limited propensity for progression to mycosis fungoides as shown by the presence of monoclonal T-cell population in a restricted T-cell profile by TCR-β gene analysis [239]. However, other studies have shown that T lymphocytes in PLC are polyclonal [229]. Immunohistochemical studies demonstrated that PLEVA is a disorder of cytotoxic T lymphocytes. The cytotoxic T cell population often presents in close proximity to epidermal keratinocytes, suggesting that the primary event is epidermal damage induced by CD8+ cytotoxic T cells [236].
Although PLC is believed to be a T-cell disorder, it may also be a manifestation of a hypersensitive reaction to bacterial or viral infectious agent similar to PLEVA. Parvovirus B19 has been suggested as a causative infectious agent [236, 240]. Other infectious agents have been implicated as evident by sporadic outbreaks of the disease in patients with a history of previous upper respiratory tract infection, mononucleosis, or infections with Toxoplasma gondii and human immunodeficiency virus [236, 241, 242].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


