3 Immunology of graft rejection
Introduction
The optimal treatment of end-stage renal failure is transplantation, since it enhances both quality of life and survival compared with dialysis. The combination of pancreatic and kidney transplantation has similar benefits whilst transplantation of other organs is truly life saving since no long-term biomechanical support is available.
In clinical practice, organ donor and recipient are genetically distinct except in the case of identical twins (see Box 3.1 for terminology). The genetically encoded immunologically mediated barrier to transplantation was defined over the course of the 20th century. The study of transplantation has played a pivotal role in defining fundamental immunological phenomena and immunology has provided a rationale for the development of clinical transplantation.
Box 3.1 Transplant terminology
These fundamental observations form the scientific basis of transplantation, and there is now a highly sophisticated understanding of the molecular and cellular events which lead to graft rejection or acceptance, the subjects of this chapter (Fig. 3.1).
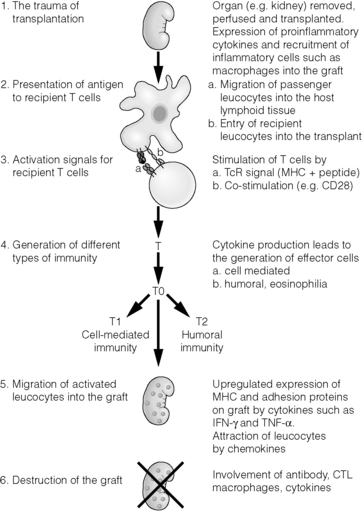
Before transplantation
Inflammation lies at the centre of the afferent and efferent arms of rejection. This begins prior to transplantation in the haemodynamic and neuroendocrine responses to brainstem death, in the process of multiorgan retrieval and the act of cold preservation. The transplant surgical procedure then adds a period of warm ischaemia and reperfusion. The consequent injury contributes to delayed graft function and generates an inflammatory response, which may itself promote and shape alloantigen-specific immunity.
The importance of these aspects of transplantation is illustrated by:
Ischaemia–reperfusion injury (IRI)
Early following reperfusion, oxidative stress induced by IRI activates a range of cell types with consequent release of immunologically active soluble proteins such as interleukins 1 and 6 (IL-1 and IL-6) and various chemokines. Vascular endothelium is activated, becoming prothrombotic and expressing adhesion molecules (selectins) that mediate local leucocyte recruitment. The expression of selectins, integrins, cytokines and chemokines promote leucocyte extravasation, via processes that include rolling, formation of tight adhesions and endothelial transmigration. There is an early infiltrate of inflammatory cells even in syngeneic transplants in which alloimmunity is absent. IL-6 released systemically during reperfusion may suppress the generation of regulatory cells (see below), leading to further augmentation of a damaging response.
The severity of this initial injury and the nature of the subsequent inflammatory infiltrate is likely to be important in the stimulation of specific alloimmunity: a maximally damaged organ generates a maximal ‘danger signal’1 which initiates productive immunity manifest as rejection. The relationship between cellular infiltration and outcome is not simple in that this also occurs during the acquisition of tolerance in many animal models of transplantation. The context of such an initial encounter with alloantigen must therefore be important in determining the subsequent nature of any response. The mechanisms by which innate immunity shapes the ensuing responses to alloantigen are becoming increasingly defined. These include Toll-like receptor (TLR) engagement and γ-interferon (γ-IFN) production by natural killer (NK) cells activating dendritic cells (DCs). TLRs expressed by antigen-presenting cells (APCs) are a family of pattern recognition receptors that recognise microbes and endogenous stress proteins such as heat shock proteins whose ligation leads to APC maturation.2 TLR activation and upregulation has been demonstrated in models of IRI.3 The relationship between TLR engagement and acute allograft rejection has also been shown in clinical studies of lung and kidney transplantation.4 IRI also activates other aspects of innate immunity such as complement that may contribute to immediate graft damage and influence adaptive immune responses.
Histocompatibility
Although differences in multiple minor histocompatibility antigens alone can result in allograft rejection in rodents, in clinical solid-organ transplantation the MHC is of overwhelming importance. However, this is not the case in bone marrow transplantation in which grafts between human leucocyte antigen (HLA)-identical siblings may be rejected or cause graft-versus-host disease5 due to differences in only a limited number of minor antigens.
Antigens stimulating rejection by T lymphocytes
MHC class I and II
The MHC is divided into three regions, namely the class I, II and III regions, and these are described in Chapter 4. A large array of genes has been localised to the MHC by sequencing, but two major classes of transplantation antigens were those first mapped by classical genetic techniques. The structure of these molecules is shown in Fig. 3.2.
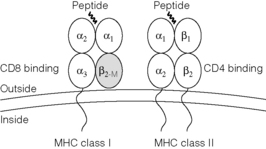
MHC class I proteins are cell-surface glycoproteins comprising a heavy chain (≈︀45 kDa), which is highly polymorphic, and a less variable light chain, β2-microglobulin (≈︀12 kDa), which is encoded outside of the MHC. β2-Microglobulin is not anchored in the membrane and binds non-covalently to the heavy chain. MHC class I proteins are expressed on most nucleated cells and generally engage T lymphocytes bearing the cell-surface protein CD8. CD8 is required for effective TCR engagement of MHC class I, resulting in intracellular signalling.
Structure of MHC class I and II proteins
MHC class I and II proteins have a similar three-dimensional structure forming a ‘peptide binding groove’ between two alpha helices (Fig. 3.2). The amino acids that constitute this groove are those which vary most between allotypes. During synthesis and transport of MHC class I and class II proteins to the cell surface the groove binds a wide range of short peptides. The groove in MHC class I has closed ends and the peptides bound are therefore 8–10 amino acids long. The groove in MHC class II is open and can accommodate peptides of considerably greater length. The sequences flanking those bound within the groove whilst not contributing directly to antigen specificity may contribute to the physicochemical properties of the complex. The peptides bound to MHC class I are primarily derived from intracellular proteins (but see ‘Indirect/cross-presentation’ below) and those from MHC class II primarily from extracellular proteins (Fig. 3.3). This difference arises from their distinct intracellular trafficking. In the presence of infection, foreign protein is processed to generate peptides bound to MHC forming a compound antigenic determinant, which engages the TCR and stimulates T-lymphocyte activation. This compound determinant is the structural basis for self-restricted antigen recognition by a T-cell repertoire that is skewed, by thymic selection, toward the engagement of peptide presented in the context of self-MHC. In the absence of foreign protein, all peptides are derived from self-proteins to which the individual is, to varying degrees, tolerant. A significant proportion of self-peptides are derived from MHC proteins themselves and this has consequences for allorecognition.
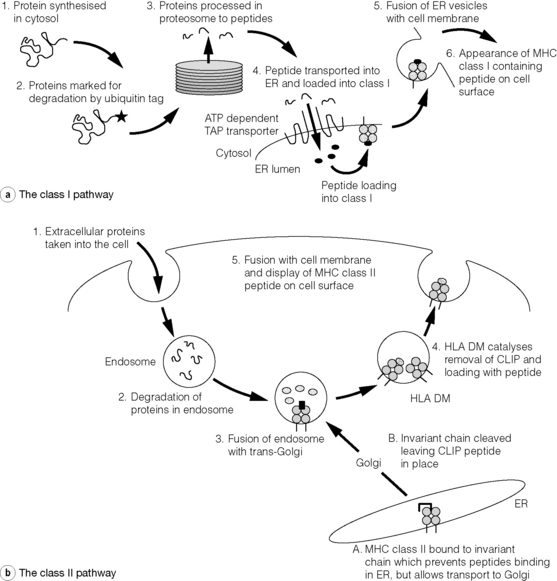
Figure 3.3 Antigen processing and presentation in the MHC class I and II pathways. (a) Processing of endogenous antigens occurs primarily via the class I pathway. Peptides are produced and loaded into MHC class I proteins as shown in steps 1–4. During the synthesis of MHC class I proteins (steps 1–3) the α chain is stabilised by calnexin before β2-microglobulin binds. Folding of the MHC class I/β2-microglobulin remains incomplete but the complex is released by calnexin to bind with the chaparone proteins, tapaisin and calreticulin. Only when the TAP transporter delivers peptide to the MHC class I/β2-microglobulin can folding of this complex be completed and transport to the cell membrane occur (steps 5 and 6). (b) Processing of exogenous antigens occurs primarily via the class II pathway. Antigens are taken up into intracellular vesicles where acidification aids their degradation into peptide fragments (steps 1 and 2). Vesicles containing peptides fuse with trans-Golgi, containing CLIP–MHC class II complexes (step 3). HLA-DM aids removal of CLIP and loading of peptide before the class II peptide complex is displayed on the cell surface (steps 4 and 5). MHC class II proteins are synthesised in the endoplasmic reticulum, where peptide binding is prevented by invariant chain. Invariant chain is cleaved leaving the CLIP peptide still in place (steps A and B) before fusing with acidified vesicles containing peptide. In B lymphocytes and epithelial cells of the thymus an atypical class II protein, HLA-DO, is expressed; this is a dimer of HLA-DNα and HLA-DOβ. It, like HLA-DM, is not expressed at the cell surface and inhibits the action of HLA-DM. Its precise role is unknown. ATP, adenosine triphosphate; CLIP, class II-associated invariant chain peptide; ER, endoplasmic reticulum; MHC, major histocompatibility complex; TAP, transporters associated with antigen processing.
The specific structure of the binding groove determines the peptide that can be bound by any given MHC and there are a limited number of peptides from a given protein that form a stable complex. The ontogenetic drive for extreme polymorphism in the MHC is therefore likely to reflect the need to counter a wide variety of infectious organisms that rapidly mutate and could evolve protein sequences that do not bind an individual’s MHC repertoire. This is unlikely to occur across a whole species if there is a full range of polymorphism as observed in humans. Certain species with limited polymorphism at either MHC class I or II can be devastated by infections that, in closely related species with polymorphic MHC, do not threaten the population.
Assembly of the MHC–peptide complex
The assembly of the MHC–peptide complex (Fig. 3.3) involves sophisticated mechanisms of antigen uptake, processing to peptide and, in the case of MHC class I, transport into the endosomal compartment. The proteosome components (low-molecular-mass polypeptide, LMP) and transporters associated with antigen processing (TAP) responsible for these aspects of processing are encoded within the MHC. In the case of classical MHC class II, assembly requires the initial presence of an invariant chain from which a peptide, class II-associated invariant chain peptide (CLIP), combines with α and β chains to form a nascent complex. CLIP is subsequently exchanged for antigenic peptide.
Non-classical MHC
Molecules such as HLA-DM, TAP and LMP that have a role in antigen processing are encoded within the MHC locus as are other molecules more broadly involved in immunity, for example tumour necrosis factors α and β (TNF-α and TNF-β) and complement components C2 and C4. Polymorphisms linked to these genes have been implicated in disease expression and transplantation responses.6
HLA-E and HLA-G are expressed in the placenta and are thought to play a role in modulating alloimmune responses to the fetus. There is emerging evidence that the expression of these molecules may also be associated with improved outcome in allogeneic transplantation.7,8 HLA-E modulates alloimmunity through its interaction with CD94-NKG2A, an inhibitory receptor expressed on NK and cytotoxic T cells. HLA-G interacts with inhibitory receptors ILT-2 and -4 expressed by APCs, NK and T cells. In a large series of renal transplant patients, the formation of soluble HLA-G was strongly correlated with diminished acute rejection rates.8
MHC class I-related chain
MHC class I-related chain A (MICA) genes are located within the class I region of the chromosome and are highly polymorphic. Although constitutively expressed at low levels by various cell types, including monocytes, epithelial cells, fibroblasts and endothelial cells, they may be upregulated by stress signals of oxidative stress and sepsis. Expression of MICA in response to stress may activate NK and T cells through its interaction with cytolytic activating receptor NKG2D. MICA is polymorphic and its alloantigenicity has been demonstrated in animal models. There is now good evidence that formation of antibody to MICA is associated with poor graft outcomes in renal transplantation9 and increased rates of acute rejection in cardiac transplantation.10
Minor histocompatibility antigens
The crystallographic structure of the MHC with bound peptide provides a structural explanation for these properties of miH antigens. They are peptides derived from proteins of limited polymorphism that, when bound to syngeneic MHC, constitute an antigenic determinant in the same way as any conventional antigen. This explains why it is difficult to raise antibodies to miH antigens, since antibodies typically engage conformational determinants on proteins and the B-lymphocyte receptor is less discriminating of subtle differences in the structure of peptide–MHC composites than is the TCR.
The most easily defined miH antigen is the male-specific H-Y antigen, which is in fact a series of antigenic peptides derived from proteins encoded on the Y chromosome, accounting for its consistent detection across a wide range of background MHC (although in certain strain combinations there is a single dominant antigen11). In rodent studies the role of miH proteins as transplantation antigens can be readily demonstrated, particularly when there has been prior sensitisation to the relevant antigen.
In rare cases of rejection in renal transplants between HLA-identical siblings, miH are thought to be the inciting antigens but their importance in other forms of solid-organ transplantation is generally limited. This is not true, however, in bone marrow transplantation, in which rejection is easily stimulated by such differences causing graft vs. host or graft vs. tumour response.12
Recognition of alloantigen
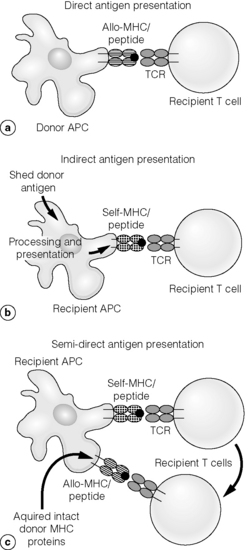
Direct allorecognition
Allogeneic MHC on DCs derived from the graft will be occupied by any number of different endogenous peptides (Fig. 3.4). The frequency of alloreactive T lymphocytes stimulated through this pathway is high, of the order of 1/102. Although this appears to contradict the principles of ‘self-MHC’ restriction resulting from thymic education, it emerges as a consequence of the vast array of endogenous peptides that occupy the MHC generating an equivalent number of compound epitopes. It also arises from the relatively limited differences in the structure of MHC alloantigens outside the peptide-binding groove and the corresponding fact that T lymphocytes can distinguish subtle differences in the kinetics of TCR–peptide–MHC interactions.
These facts apply equally to the response to recall antigens; that is, other than in naive animals any direct alloresponse will include cross-reactions with self-restricted secondary responses to conventional antigens.13–16 As most studies on experimental transplantation, particularly those on the induction of tolerance, use naive rodents these are unlikely to accurately reflect the alloresponse observed clinically. Indeed, strategies to induce transplantation tolerance which succeed in the naive animal are unsuccessful in previously infected animals,15,16 providing one possible explanation for the difficulties in translating such strategies into preclinical models.
Indirect allorecognition
The potential importance of this route of allorecognition is demonstrated by experiments in which immunisation with MHC class I peptides prime for subsequent rejection of a skin allograft bearing the intact class I antigens from which the peptides were derived.17 Studies in other models have demonstrated that priming with MHC class I- and II-derived peptides lead to chronic allograft rejection, with accelerated vasculopathy.18,19 These peptides are most likely to be presented in the context of recipient class II since they are extracellular for the self-APCs. However, there is now evidence of crossover or ‘cross-presentation’ between the two pathways, so there may also be presentation of such peptides in the context of MHC class I.20,21
The importance of indirect presentation of alloantigen is also illustrated by transplantation of skin allografts from MHC class II deficient onto normal mice. Donor APCs are unable to stimulate recipient CD4+ T cells and yet rejection remains dependent upon CD4+ T cells. It is likely that this reflects recipient CD4+ T-cell stimulation through indirect presentation of donor alloantigens by self-MHC.20–22 In this latter case, of course, peptides are not artificially introduced into the recipients but produced by normal processing of MHC from apoptosed donor-derived cells.
In long-standing renal transplant recipients there is evidence of donor-specific hyporesponsiveness of peripheral blood lymphocytes to directly presented alloantigen,27–29 and this may be true even in those who have suffered allograft failure.27 There is increasing evidence, however, for the association of indirect pathway alloreactivity in peripheral blood lymphocytes with chronic rejection in solid-organ transplants.28,30–32 The significance33 and interpretation of peripheral responses as indicative of tissue responses must be guarded, but these observations suggest strategies for monitoring patient responses32 and assessing the effects of treatment34 for chronic rejection.
Semi-direct allorecognition
If host T cells are stimulated by recipient-derived DCs via indirect antigen presentation, the MHC restriction of the effector cell population will be to host rather than donor. A problem could arise if a cytotoxic T cell, once stimulated with self-MHC and allogeneic peptide, comes to lyse its target cell – in the case of graft rejection, the foreign transplanted tissue, which does not express self-MHC molecules. This problem is overcome if the foreign MHC on the target cell cross-reac with self insofar as the T cell is concerned, if the effector arm of the immune response does not require MHC restriction (e.g. macrophages, DTH) or if the effector population is primed by the donor-derived MHC. How could this latter situation arise if the T cells are primed by recipient-derived DCs? It has been known for several years now that intact proteins can be exchanged between cells in cell culture systems and indeed that MHC proteins transferred in this fashion can stimulate alloreactive responses.23–25 The importance of this in stimulation of alloreactive responses in the whole animal has been highlighted in recent work,26 although its importance in inducing graft rejection has yet to be established.
T-cell interactions with antigen-presenting cells: initiation of the immune response
‘Passenger leucocytes’ in transplantation
In rodent models of transplantation, allogeneic MHC can be more or less immunogenic according to the context in which it is encountered by the recipient immune system. Following transplantation, ‘passenger leucocytes’ (tissue-resident immature DCs) migrate to secondary lymphoid organs, mature and there deliver a powerful stimulus to the recipient immune system (direct allostimulation). Mature DCs express high levels of MHC class I and II and can therefore stimulate both CD8+ and CD4+ T lymphocytes. They also have other properties including efficient co-stimulatory activity, which render them uniquely powerful stimulators of naive T cells35–37 and earn them the title ‘professional’ APCs. A number of experimental systems have been used to dissect the role of donor DCs in stimulating allograft rejection.
Yet, clear data implicating the DC as the primary (or only) stimulus of graft rejection in all donor–recipient pairs is lacking. Also, direct application of these findings to clinical transplantation must be cautious, given the fact that in many experimental models of long-term graft survival an active regulatory immune response plays a role in the failure of organ rejection. DCs themselves may be important for the induction of a regulatory response rather than rejection, although this may be most relevant to recipient DCs (indirect pathway) rather than donor DCs (direct pathway). Furthermore, although DCs are very important in the stimulation of the primary T-cell response, they seem less important for secondary responses. In clinical transplantation a significant proportion of the alloresponse is likely to be heterologous13,15 – that is, primed, self-restricted, pathogen-specific, T-lymphocyte cross-reacting on an allogeneic MHC–peptide complex. These primed cells are significantly less dependent upon DCs for activation. Nevertheless, the stimulation of lymphocytes through the ‘direct’ pathway of allorecognition by the migration of ‘passenger’ DCs seems often to provide a powerful early stimulus to acute graft rejection,38,39 whether this is a primary or secondary response.
Vascular endothelium constitutively expresses MHC class II in humans and it too may play a greater role in stimulating alloimmunity than in rodents, offering a further explanation for the difficulties of translating animal models of transplantation tolerance to the clinic.40,41
Activation of dendritic cells
These set DC phenotype, which may then determine the phenotype of the subsequent adaptive immune response. Generally ‘immature’ DCs play a role in maintaining peripheral tolerance by both deletional and regulatory mechanisms, and these properties may themselves be regulated by T lymphocytes. DCs therefore bidirectionally connect innate and acquired immunity, and by doing so link the nature of antigen-specific memory to the context in which antigen is first encountered.42,43
Co-stimulation
Engagement of the TCR can result in a range of outcomes depending upon context, as already alluded to in the discussion of DCs. This context is established by the engagement of co-stimulatory molecules on APCs and soluble mediators including cytokines. The outcome for the T lymphocyte includes proliferation, apoptosis or anergy, acquisition of different effector phenotypes, acquisition of different regulatory phenotypes or differentiation into a memory cell.
Co-stimulatory interactions can be broadly separated into those of the CD28–B7 family and those of the tumour necrosis factor receptor (TNFR)/tumour necrosis factor (TNF) family, as summarised in Table 3.1. The relative importance of different co-stimulatory pathways and the way in which they interact remains to be fully elucidated. In alloimmunity it is not yet clear whether different pathways are functionally redundant or, more likely, that they play specialised roles relevant to different aspects of successful transplantation.
Table 3.1Members of the CD28 and CD40–ligand (CD154) families of molecules expressed on T lymphocytes and their corresponding ligands
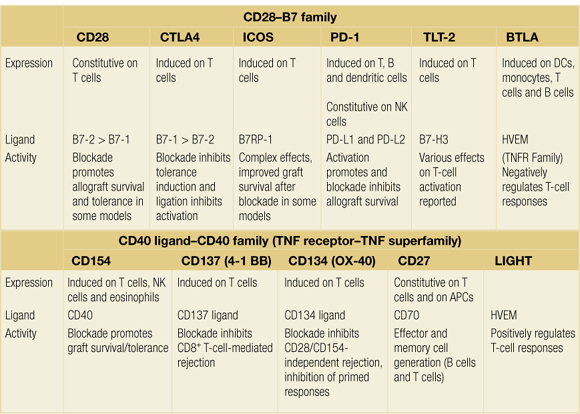
CD28–B7
The role of CD28 has been the most intensively investigated in the field of co-stimulation.44–46 CD28 is a homodimeric glycoprotein present on the surface of T cells, which interacts with two counter- receptors, CD80 and CD86, expressed on the surface of APCs. CD86 is expressed constitutively at low level by APCs and is upregulated rapidly following interaction with the T lymphocyte. It has a rather low affinity for CD28 whereas CD80, which is not constitutively expressed, is upregulated with slower kinetics but has an approximately 10-fold greater binding affinity for CD28 than does CD80. The result of ligation of CD28 by either CD86 or CD80 appears to be increased cytokine synthesis and proliferation, and they do not appear to be qualitatively distinct. The different kinetics of expression and affinity for other ligands such as CTLA4 (CD152) may result in different effects on cellular phenotype.
CD28 ligation by CD80/86 promotes initial T-cell activation but also plays a role in CTLA4 (CD152) regulatory T-cell homeostasis. Blocking the CD28 pathway in rodents can have dramatic effects on the generation of primary immune responses and may result in prolonged graft survival or even tolerance of grafts in some experimental models,47,48 but mice with a disrupted CD28 gene can make productive immune responses, albeit with sometimes altered kinetics. This may be due to redundancy of co-stimulatory pathways, including other members of the B7 and CD28 families.
CTLA4 ligation by CD80/86 inhibits T-cell activation. The severe phenotype of CTLA4 −/− mice, which die from uncontrolled lymphoid proliferation shortly after birth,49 suggests that there is less redundancy in this aspect of immune regulation.
The most widely used method to block CD28–B7 interactions has been through the use of CTLA4-Ig but this will also block CTLA4–B7 interaction, and inhibition of negative CTLA4 signalling may account for diverse observations on immune responses in different experimental systems. A high-affinity analogue of CTLA4-Ig has been developed as an immunosuppressive for use in clinical practice,50 and the development of reagents that preferentially inhibit the CD28–B7 interaction is also ongoing.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


