, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
26.1 Traumatic Neuroma
26.1.1 Clinical Features
The exact incidence of traumatic neuromas is not known, but they appear to be the most common subtype of cutaneous neuromas described [1, 2]. They may occur following any variety of injuries and at any age, including neonates and young infants, as a result of in utero amputation of a supernumerary digit [3]. Traumatic neuromas are skin colored to slightly erythematous, intermittently painful nodules, occurring at sites of previous trauma, most commonly on the digits [1, 2]. Neuromas are almost universally painful and persistent. Surgical excision may be curative.
26.1.2 Histology
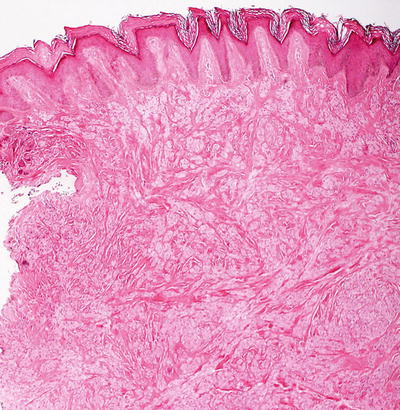
The histologic findings are identical in traumatic neuroma and amputation neuroma secondary to polydactyly. The findings are confined to the dermis, in which there is a proliferation of nerve bundles that are readily apparent at low magnification (Fig. 26.1). Large bundles of sharply circumscribed neural elements are present throughout the dermis and in the subcutis. The neural elements do not have cytologic atypia and mitotic activity is not present. Perineurial sheaths confine each bundle. Concentric rings of fibrosis resembling onion skins surround each of the nerve fascicles and are responsible for the characteristic histologic appearance (Fig. 26.2).
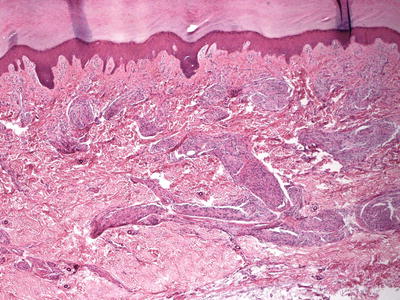
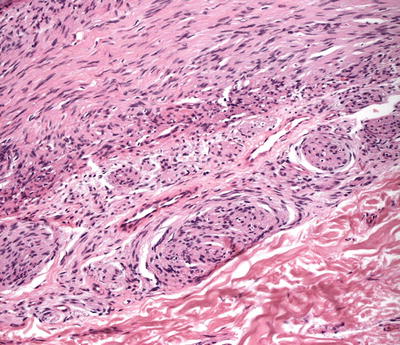
Fig. 26.1
A traumatic neuroma demonstrates multiple foci of neural proliferations throughout the dermis
Fig. 26.2
When cut en face, the neural proliferations may resemble onion bulbs in a traumatic neuroma
26.1.3 Pathogenesis
Traumatic neuroma represents a hyperplastic response of the affected nerve to injury, trauma, or chronic inflammation [4, 5]. It is believe to be secondary to dysregulated proliferation of the nerve and connective tissue in the process of reinnervation of the affected area. The development of traumatic neuroma appears to be dependent on local neurotropic molecules, particularly nerve growth factor (NGF) [6–8]. NGF is an important neurotropic factor that is essential for the survival of peripheral neurons [9]. Studies have shown that inhibition of NGF activity by blocking NGF receptor can prevent traumatic neuroma formation in rodents [7].
26.2 Neurofibromatosis Type 1
26.2.1 Clinical Features
Neurofibromatosis type 1 (NF1) is a progressive multisystem disease, secondary to genetic mutation of the NF1 gene on chromosome 17, which encodes the tumor suppressor protein, neurofibromin. The disorder has a prevalence of 1 per 3,500 people worldwide with autosomal dominant inheritance pattern, although approximately 50 % of cases occur as a result of spontaneous mutation [10]. There is significant phenotypic variability among affected individuals, but almost all patients have stigmata of disease by 20 years of age. Cutaneous diagnostic criteria include more than six café au lait macules, neurofibromas, intertriginous freckling, and plexiform neurofibroma. Additional clinical features that are commonly seen include learning disabilities, Lisch nodules, optic gliomas, and osseous lesions, such as scoliosis and sphenoid wing dysplasia. Studies indicate that the life expectancy of individuals with NF1 is lower than that of healthy individuals with increased mortality secondary to early onset vasculopathy and soft tissue malignancies, particularly malignant peripheral nerve sheath tumors [11]. The histology and pathogenesis of neural tumors in the skin that are commonly associated with NF1 are described elsewhere in this chapter (Sects. 26.4 and 26.5).
26.3 Neurofibromatosis Type 2
26.3.1 Clinical Features
Neurofibromatosis type 2 (NF2) has an incidence between 1 per 33,000 and 1 per 87,000 individuals [12]. Fifty percent of cases are attributed to spontaneous mutation of the NF2 gene region of chromosome 22, while the remainder is inherited in an autosomal dominant manner [13, 14]. Only 20 % of cases are diagnosed in children. Small café au lait macules may be present, but they are fewer in number than those seen in NF1. Multiple cutaneous schwannomas are a well-established feature of NF2 [13, 14]. The schwannomas of NF2 frequently are slightly pigmented plaques with overlying hypertrichosis. Adults frequently present with tinnitus, hearing loss, and problems with balance secondary to acoustic schwannomas. Eighty percent of children with NF2 have cataracts at the time of diagnosis as an initial sign of disease, preceding hearing loss in most cases [13]. NF2 in children portends a worse prognosis with greater mortality secondary to increased number of rapidly progressive central nervous system tumors. The histology and pathogenesis of neural tumors in the skin that are commonly associated with NF2 are described elsewhere in this chapter (Sects. 26.4 and 26.5).
26.4 Neurofibroma
26.4.1 Clinical Features
Solitary or isolated neurofibromas are relatively common benign tumors of neuromesenchymal tissue that are seen equally in both males and females. Lesions rarely occur in infancy, and more frequently present in middle to late childhood after puberty [11]. Four classifications exist: (1) discrete cutaneous neurofibromas, (2) discrete subcutaneous neurofibromas, (3) nodular plexiform neurofibromas, and (4) diffuse plexiform neurofibromas.
Neurofibromas typically present as solitary, soft, skin-colored nodules with a variable diameter between 0.2 and 2 cm in size. Lesions grow slowly and can become more exophytic or pedunculated over time [11]. A characteristic feature is invagination into the dermis when direct pressure is applied on top of the lesion, known as the “buttonhole sign.” These are benign lesions and can be treated with simple excision. Patients with multiple lesions or other cutaneous stigmata (specifically café au lait macules and intertriginous freckling) warrant evaluation for neurofibromatosis.
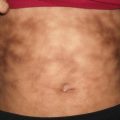
Plexiform neurofibromas are subcutaneous masses or plaques, often with overlying hyperpigmentation and a rubbery consistency on palpation likened to a “bag of worms” (Fig. 26.3). These lesions are pathognomonic of neurofibromatosis type 1, and have a reported risk of malignant degeneration in 2–5 % of cases [15].
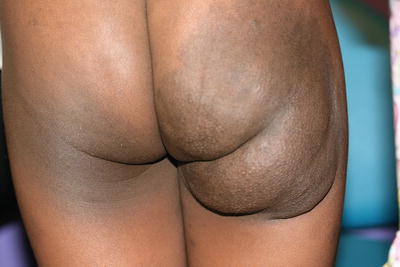
Fig. 26.3
Plexiform neurofibroma presents as a large brown patch overlying a soft subcutaneous mass on the right buttock
Malignant peripheral nerve sheath tumor is an uncommon neural malignancy with an estimated incidence of 0.0001 % in the general population [16]. Patients with neurofibromatosis type 1 (NF1) share a greater burden of disease with incidence of 1 case per 100 NF1 patients per year [16]. Tumors typically present in middle-aged adults, although median age of onset in those with NF1 is younger (mid-30s), including reports of diagnosis in adolescents. Lesions commonly occur in the setting of nodular and plexiform neurofibromas, and present as painful, expanding masses. Poor prognostic factors include tumor size at the time of diagnosis and axial location. Surgical excision is the standard of care as tumors have poor response to radiotherapy and chemotherapy. Ultimately, the diagnosis carries a poor prognosis with a 50 % 5-year survival rate in those without NF1 and 15 % in those with the disease [16].
26.4.2 Histology
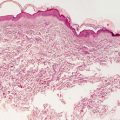
Histologic findings in neurofibromas are those of a spindle cell proliferation based in the dermis (Fig. 26.4). The epidermis is either unremarkable or somewhat flattened when the underlying neoplasm extends higher in the dermis. Occasional cases demonstrate changes within the epidermis and appendages [17]. The tumor consists of a population of spindle-shaped cells with tapered nuclei. The surrounding stroma is characteristically pale staining, especially when compared with surrounding dermis (Fig. 26.5). In some cases, the stroma may have foci of myxoid change. The tumor is relatively well-circumscribed and not encapsulated. Diffuse neurofibromas that are not at well-circumscribed are more prevalent in children [18]. Occasional cells demonstrate cytologic atypia in some cases with this appearance usually being attributed to “ancient change.” When the cytologic atypia is more pronounced, a diagnosis of atypical neurofibroma may be warranted [19]. Mitotic activity is rare, and should raise concern for malignancy when abundant. Similarly, marked increase in cellularity should raise suspicion of malignant peripheral nerve sheath tumor [19]. Most cases do not have a significant inflammatory response, but mast cells are present in neurofibromas. Admixed adipocytes may be present as either diffuse or focally dispersed throughout the lesion.
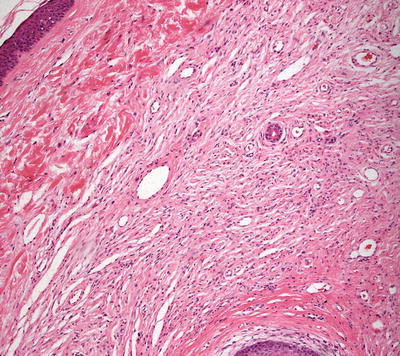
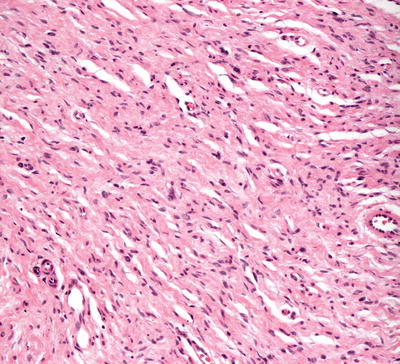
Fig. 26.4
Unencapsulated spindle-shaped cells coursing in pale eosinophilic neuropil characterize neurofibroma
Fig. 26.5
The cells in neurofibroma as wavy and thin with tapered ends
Plexiform growth pattern can be seen in the skin with several separate lobular configurations, but extreme caution is advised regarding making the diagnosis of plexiform neurofibroma on a skin biopsy. This diagnosis confers a diagnosis of neurofibromatosis, and gross examination of lesions and genetic testing are the preferred ways to establish this diagnosis. Rare variants may contain nests of nonpigmented melanocytes, and are designated cutaneous epithelioid melanocytic neurofibromas [21]. It is unclear whether these tumors represent a largely neurotized melanocytic nevi. Another variant known as a pigmented storiform neurofibroma demonstrates a storiform growth pattern and scattered pigment-laden cells [22].
The differential diagnosis includes dermatofibroma. These tumors usually demonstrate overlying epidermal hyperplasia and hyperpigmentation, which are features not associated with neurofibroma. Furthermore, the interspersed collagen tends to be deeply eosinophilic as opposed to the pallor of the stroma in neurofibromas. Neuromas consist of a more lobulated growth pattern with individual nerve fascicles surrounded by perineurium. These findings are not seen in neurofibromas. Schwannomas demonstrate palisading as well as Antoni A and Antoni B areas with marked differences in cellularity, which are features not seen in neurofibroma. Moreover, Schwannomas are encapsulated unlike neurofibromas which are not.
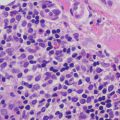
Malignant peripheral nerve sheath tumors are very uncommon in children but can arise in patients with NF1 [23, 24]. In most cases, the tumors appear to arise from within a plexiform neurofibroma. A proliferation of spindle-shaped cells with tapered nuclei and relatively scant cytoplasm is present within the dermis, growing in a fascicular and infiltrative pattern (Fig. 26.6). These tumors are markedly hypercellular with abundant pleomorphism, cytologic atypia, and mitotic activity (Fig. 26.7) [19]. Single cell necrosis and areas of zonal necrosis help to identify this as a malignant neoplasm.
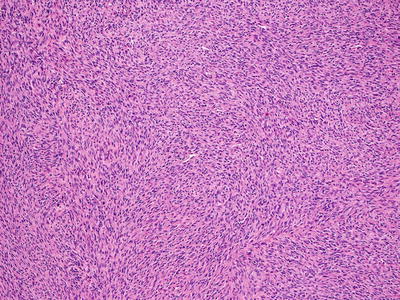
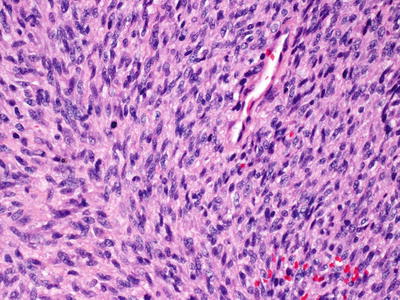
Fig. 26.6
Malignant peripheral nerve sheath tumor demonstrates sheets of wavy cells coursing in fascicles
Fig. 26.7
Malignant peripheral nerve sheath tumor demonstrates pleomorphism and cytologic atypia
The differential diagnosis includes monophasic synovial sarcoma, leiomyoma, melanoma, and dermatofibrosarcoma protuberans. Immunohistochemistry is helpful in making the diagnosis as well as genetic testing of the tumor (Fig. 26.8) [25]. Melanotic schwannoma also enters the differential diagnosis, but it can be distinguished based upon the abundant pigment within the tumor and its Schwannian growth pattern [26].
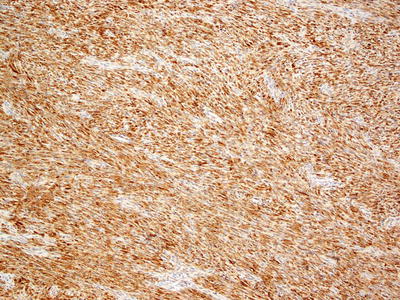
Fig. 26.8
Diffuse S100 staining helps to differentiate malignant peripheral nerve sheath tumor from other spindle cell neoplasms that may occur in the dermis
26.4.3 Pathogenesis
Neurofibroma is a benign peripheral nerve tumor that is composed of Schwann cells, perineurial cells, axons, fibroblasts, and mast cells [27]. Neurofibroma is likely to arise from Schwann cell lineage [28]. Mast cells can promote neurofibroma growth, and fibroblasts and endothelial cells can enhance angiogenesis and collagen synthesis in neurofibroma [29, 30]. Cutaneous neurofibroma is a benign growth, and does not undergo malignant transformation, unlike plexiform neurofibroma, which can be associated with transformation to malignant peripheral nerve sheath tumor [31, 32]. Neurofibroma can occur sporadically or in the setting of neurofibromatosis type 1.
The RAF/MEK/ERK signaling pathway has a key role in the growth of neurofibroma and malignant peripheral nerve sheath tumor [33]. Inhibitors of MEK effectively reduce the growth of neurofibroma in vitro and in experimental animal models [32]. There are currently active clinical trials of MEK inhibitors for patients with neurofibromatosis-associated neurofibromas and plexiform neurofibromas [32, 34].
26.5 Schwannoma
26.5.1 Clinical Features
Cutaneous schwannomas are rare and may occur at any age, but lesions most frequently are described in adults [35]. Cutaneous schwannomas typically are asymptomatic soft, pink to yellow, smooth and deep-seated subcutaneous nodules or plaques of the head and neck with diameter between 0.5 cm and 3 cm in size [35, 36]. They can sometimes be painful. Lesions are usually solitary, but descriptions of multiple schwannomas have been reported in familial cases, and in association with genodermatoses, such as neurofibromatosis type 2 and neurilemmomatosis (when other features of neurofibromatosis type 2 are not seen) [13, 14]. Schwannomas are benign nerve sheath tumors, and can be treated with simple excision [35].
26.5.2 Histology
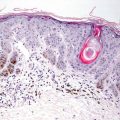
Cutaneous schwannomas share histologic features with those occurring in other organs. They are a well-circumscribed and encapsulated proliferation of neural elements in the dermis. Cellular areas (Antoni A) and paucicellular areas with more myxoid matrix (Antoni B) are interspersed. Verocay bodies, which are a palisade of tumor cells, are readily identifiable in many cases (Figs. 26.9, 26.10, 26.11, and 26.12). Ancient change may account for slight cytologic atypia seen in some cases. Mitoses are not a feature of this neoplasm, and their presence should raise suspicion for malignancy. Rarely, a plexiform growth pattern has been described in the skin, most often associated with neurofibromatosis [14, 37]. This growth pattern has also been described in the rare epithelioid schwannoma variant characterized by tumor cells with an epithelioid morphology and less spindly than the usual schwannoma [38–40].
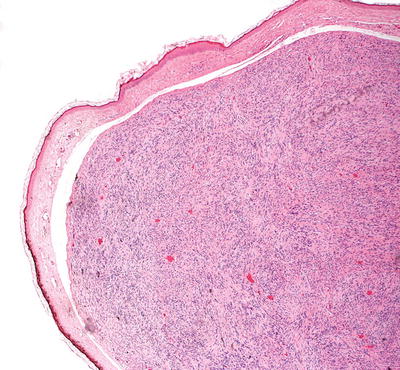
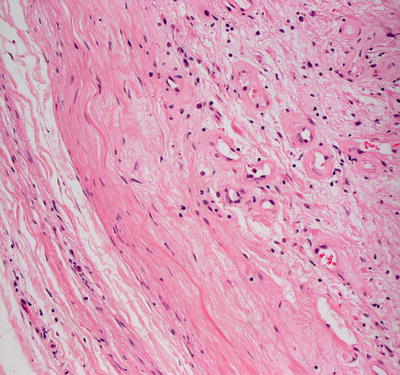
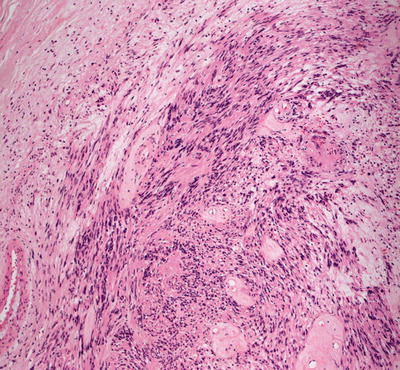
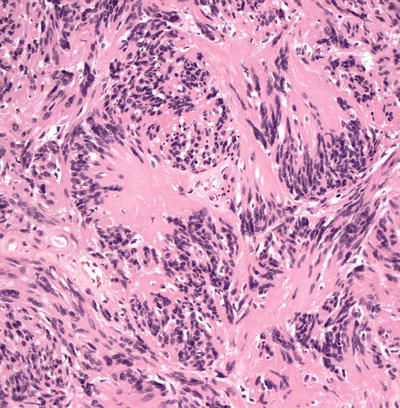
Fig. 26.9
Schwannomas are sharply circumscribed dermal tumors comprised of spindle-shaped cells
Fig. 26.10
Some areas of schwannoma may have wispy background collagen and a proliferation of small vessels
Fig. 26.11
More cellular areas of schwannomas may demonstrate a pseudopalisading pattern, giving rise to Verocay bodies
Fig. 26.12
Verocay bodies demonstrate nuclear palisades and a characteristic appearance
A variant with multiple pigment-containing cells and psammoma bodies has been reported to be a marker for Carney syndrome, and is known as a melanotic psammomatous schwannoma [41, 42]. A melanotic component in the absence of psammoma bodies does not always confer a diagnosis of Carney syndrome [26]. A pseudoglandular schwannoma demonstrates the usual characteristics of schwannoma but also has pseudoglandular spaces. There is no significance to this particular histologic variant [43]. A very rare neuroblastoma-like schwannoma variant has been described with smaller cells and rosette formations [44, 45].
The differential diagnosis includes other neural neoplasms. Neurofibromas do not demonstrate Verocay bodies and, while usually well-circumscribed, they are not encapsulated. S100 immunohistochemistry diffusely and strongly stains schwannomas, in which most of the tumor elements are axons, but only show partial S100 positivity in neurofibromas, which contain many cell types. Palisaded encapsulated neuromas may share some features with schwannomas, but they are not encapsulated and rarely demonstrate palisading (despite the name). They are also readily differentiated based upon the clinical presentation. Traumatic neuromas are characterized by a more lobulated proliferation of nerve fascicles than schwannoma.
26.5.3 Pathogenesis
Schwannoma is a benign tumor, consisting of a clonal population of Schwann cells. Most cases of schwannoma are sporadic. However, some cases are associated with radiation exposure and syndromic disorders, such as neurofibromatosis type 2 (NF2), schwannomatosis, and Carney’s complex [46–50]. Schwannoma in children is most commonly located in the skin in individuals with neurofibromatosis [48, 51].
The identification of NF2 (merlin) gene has led to a better understanding of the disease pathogenesis [52, 53]. Mutations in NF2 are found in sporadic and NF2-associated schwannomas, many of which are de novo mutations [48, 54]. The phosphorylation state of merlin is important for the mechanism of the loss of function and inactivation of merlin in schwannoma [55].
Schwannomas can occur as multiple lesions in some individuals (schwannomatosis). Germ-line mutations in SMARCB1 have been reported in a number of patients with schwannomatosis [56, 57]. SMARCB1 has tumor suppressor functions, and forms part of an adenosine triphosphate (ATP)-dependent chromatin-remodeling complex that is important in central nervous system development [58]. It has been found that germ-line loss of function mutations of another tumor suppressor gene called LZTR1 is common in patients with schwannomatosis. LZTR1 is known to interact with proteins that regulate SMARCB1 expression [59].
26.6 Granular Cell Tumor
26.6.1 Clinical Features
Granular cell tumor is a rare neoplasm, primarily affecting adults between 30 and 60 years of age, with rare reports of such tumors in children [60]. They occur twice as frequently in females, and are more commonly seen in African-Americans [61]. Granular cell tumors usually present as an asymptomatic solitary, skin-colored or brownish red, firm subcutaneous nodule with diameter between 0.5 cm and 3 cm in size [60]. They may occur anywhere on the body, but 45–65 % of cases present at the head or neck with most lesions described as intraoral tumors, generally affecting the tongue [62]. Patients sometimes experience associated mild tenderness or pruritus. Malignant transformation with local infiltration or metastasis occurs in approximately 2 % of cases [61]. Complete excision is recommended for symptomatic or rapidly enlarging lesions since the likelihood of recurrence is high when tumors are not completely removed.
26.6.2 Histology
Marked pseudoepitheliomatous hyperplasia is seen overlying many cases of granular cell tumors. One such tumor occurring in a child has been described with marked overlying hypertrichosis [63]. The dermis is filled with a proliferation of large cells with abundant granular and eosinophilic cytoplasm (Figs. 26.13 and 26.14). The larger cytoplasmic granules may be surrounded by a halo, in which case they are given the name pustulo-ovoid bodies of Milian. There is no significance to this finding other than recognition of this feature in some granular cell tumors [64]. The cell nuclei are small, dark, and uniform in appearance. Nucleoli are inconspicuous. Cytologic atypia is absent or minimal in most cases, and the mitotic rate is low. The tumor may be locally invasive, and is not well circumscribed. Extension into the subcutis is common. Perineural invasion and extension into arrector pili muscles are present in many cases, and vascular invasion is seen in some tumors [65]. A plexiform growth pattern has been described, and this is expected given the Schwannian origin of these neoplasms [66–68].