Fig. 4.1
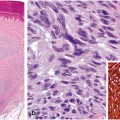
Trichilemmoma—The histology shows lobular, folliculocentric proliferations of uniform, bland basaloid cells with peripheral palisading and frequent clear cell cytoplasm (due to periodic acid–Schiff (PAS)-positive glycogen accumulation) (a). A distinctive feature is the presence of a thick eosinophilic basal membrane (b)
If solitary trichilemmomas are relatively common, multiple lesions are almost invariably associated with Cowden syndrome. Interestingly, the desmoplastic variant of trichilemmoma is not reported as being associated with Cowden syndrome [3].
Cowden Syndrome
Cowden syndrome (multiple hamartoma syndrome), originally described in 1963 by Lloyd and Dennis, is a multisystem hamartomatous disorder with associated macrocephaly, predominately affecting women [3]. The mucocutaneous manifestations of this syndrome are represented by multiple trichilemommas, predominately in the head and neck area, as well as by punctate acral keratoses of soles and hands, sclerotic fibromas, acrochordons, oral papillomas, and acrokeratosis verruciformis [4–8]. Mucocutaneous lesions are almost always identified in Cowden syndrome, are estimated to have 99 % penetrance by the 3rd decade of life, and may represent occasionally the first manifestation of Cowden syndrome [4].
Gastrointestinal polyps are present in 60–90 % of patients and benign thyroid lesions (adenomas, hamartomas, multinodular goiter, and Hashimoto thyroiditis) occur in up to 75 % of patients [9–11]. Benign breast lesions, genitourinary malformations, leiomyomas, lipomas, and vascular malformations are also described [11–13].
Most importantly, these patients have an increased risk for developing internal malignancies, mostly breast adenocarcinoma and thyroid carcinoma (usually follicular variant), gastrointestinal and genitourinary tumors, melanoma, etc [14, 15]. According to the literature reports, the lifetime risk for patients with Cowden syndrome is 85.2 % for breast carcinoma (as compared with 11 % within the general population), 35.2 % for thyroid carcinoma, 33.6 % for renal cell carcinoma, 28.2 % for endometrial carcinoma, 9 % for colonic carcinoma, and 6 % for melanoma [15]. As with other hereditary breast cancer syndromes, the breast adenocarcinoma in patients with Cowden syndrome occurs at a younger age (average 36–46 years) and male patients are also at risk of developing breast cancer [16].
Cowden syndrome is an autosomal dominant condition and is caused by a germline inactivating mutation in the tumor suppressor phosphatase and tensine homolog gene (PTEN) situated on chromosome 10q23.31 [17, 18]. PTEN is a tumor suppressor gene and is an important mediator of carcinogenesis in a variety of human malignancies [19–22]. PTEN is involved in apoptosis and cell cycle arrest and negatively regulates the cell survival and PI3K/Akt/mTOR pathway through its phosphatase activity. PTEN phosphatase uses Akt-activator phosphatidylinositol-3,4,5-triphosphate (PIP3) as a substrate [17, 21–23]. Moreover, PTEN negatively affects the mitogen-activated protein (MAP) pathway. It has been demonstrated that 80–85 % patients with Cowden syndrome have germline loss-of-function PTEN mutation, with approximately 40–60 % of cases estimated to be familial [24]. Frameshift, nonsense, and missense point mutations, deletions, insertions, and splice site mutations have been described. It was reported that although these mutations may be identified almost in the entire length of PTEN gene (with exception of first, fourth, and last exon), approximately half of the mutations were identified in exon 5, coding for the core phosphatase domain, which is responsible for phospholipid binding. Interestingly, a correlation was observed between the presence of PTEN mutation and breast adenocarcinoma [24]. Promoter mutations were associated with breast cancer, whereas colorectal carcinoma was associated with nonsense mutations .
Other mechanisms of PTEN loss-of-function, such as PTEN promoter hypermethylation (which leads to underexpression of PTEN) or PTEN inactivation through ubiquitin protein-mediated degradation, may account for the remainder cases [25]. Recently, PI3KCA and Akt1 germline mutations were reported in some patients with Cowden syndrome lacking PTEN mutations [26]. Also, germline epigenetic regulation of KILLIN was described in a subset of patients with Cowden or Cowden-like syndrome lacking germline PTEN mutations [27, 28]. KILLIN is identified as a TP53 regulated inhibitor of DNA synthesis through S phase arrest, is transcribed in the opposite (i.e., antisense) strand relative to PTEN, and shares the same promoter region as PTEN. It has been reported that individuals with KILLIN-promoter hypermethylation have a threefold increased prevalence in breast cancer and a greater than twofold increase in kidney cancer prevalence over individuals with germline PTEN mutations [28].
As a practical approach for a patient in which more than one trichilemmoma was histologically diagnosed, a difference in expression of PREN by immunohistochemistry in patients with Cowden syndrome versus sporadic cases was shown recently. It is reported that complete PTEN loss in trichilemmomas by immunohistochemistry is strongly suggestive of association with Cowden syndrome, but retention of PTEN labeling does not entirely exclude the syndrome [29].
A heterogenous group of individuals with Cowden-like syndrome, but not meeting Cowden syndrome inclusion criteria such as presence of PTEN germline mutation, is increasingly recognized. There are pathognomonic diagnostic criteria for the diagnosis of Cowden syndrome, updated annually by the US National Comprehensive Cancer Network.
PTEN hamartoma tumor syndrome (PHTS) is a term encompassing subsets of several clinical syndromes with germline mutations in PTEN tumor suppressor gene. These syndromes have autosomal dominant transmission with variable phenotypic manifestations and are characterized by germline mutations of PTEN located at 10q22–23 [17, 21–23]. Cowden syndrome is the prototypic PHTS but this category also includes: Lhermitte–Duclos disease, Bannayan–Riley–Ruvalcaba syndrome, and possibly Proteus syndrome. Lhermitte–Duclos disease is characterized by dysplastic gangliocytoma of the cerebellum which leads to increase in intracranial pressure, ataxia, and seizures. Bannayan–Riley–Ruvalcaba syndrome presents with developmental delay, macrocephaly, lipomas, hemangiomas, and pigmented penile speckled macules. Proteus syndrome is a rapidly progressive disorder characterized by mosaicism, congenital malformations, tissue overgrowth, epidermal nevi, hyperostosis, and various vascular anomalies. Notably, an increased risk for malignancy was demonstrated only for Cowden syndrome in the PHTS group [17, 22].
Clinical Management
Considering the increased risk of developing malignancies in Cowden Syndrome, the most critical aspect is close and heightened cancer surveillance. As a general rule, any patient who meets the patognomonic criteria for Cowden syndrome is indicated to be referred to genetic counseling, and sequencing of PTEN should be considered [9]. There is no definite consensus regarding the patients with Cowden-like syndrome, but not meeting the inclusion criteria [4]. Screening for breast cancer is indicated to start at an earlier age (30–35 years) and includes annual mammograms and magnetic resonance imaging (MRI), but there is also an increased concern that this will lead to a high degree of false-positive data and a high yield of unnecessary procedures. Total thyroidectomy is generally recommended in patients who develop thyroid cancer because of a high risk of recurrence. The current guidelines also recommend annual endometrial biopsies for premenopausal women and endometrial ultrasound for postmenopausal women [4, 17, 22].
The development of PI3K/Akt/mTOR inhibitors is extensively studied in oncology, as this pathway promotes cellular survival and chemotherapeutic resistance in a variety of malignancies. mTOR inhibitors are especially promising in patients with Cowden syndrome [17].
Pilomatricoma (“Calcifying Epithelioma of Malherbe”)
Clinical Features
Pilomatricoma is a relatively common skin adnexal tumor with follicular origin and differentiation toward the follicular matrix, which mimics in its evolution the hair growth. It predominately affects children and young adults, more than half of the cases developing in the first 2 decades of life, and has a 3:1 female preponderance. It usually involves the head, neck, and upper extremities, and less frequently trunk and lower extremities [30–33]. Pilomatricoma presents as frequently solitary well-circumscribed, firm, slow-growing, asymptomatic, 1–3 cm in maximum dimension, deep dermal or subcutaneous nodules covered with normal skin tissue. Rarely, variants of pilomatricomas were described: perforating, exophytic, multinodular, giant, and bullous clinical types [30–33].
Histology
The histologic features of pilomatricoma depend on its stage of development. Early lesions are frequently cystic with areas resembling a follicular cyst, infundibular type, areas containing basaloid cells resembling the follicular metrical cells (hyperchromatic nuclei, high nuclear/cytoplasmic ratio, and numerous mitotic figures) and supramatrical cells with eosinophilic cytoplasm, without nuclear structures (called shadow or ghosts cells). (Fig. 4.2) As the lesion progresses, the shadow cells predominate whereas the metrical cells become less obvious. Dystrophic calcifications and metaplastic ossification are commonly seen in this stage. Late-stage lesions can occasionally imitate osteomas.
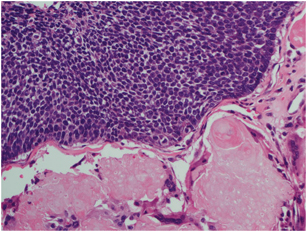
Fig. 4.2
Pilomatricoma—The histologic features reveal areas containing basaloid cells resembling the follicular matrical cells (hyperchromatic nuclei, high nuclear/cytoplasmic ratio, and numerous mitotic figures) and supramatrical cells with eosinophilic cytoplasm, without nuclear structures (shadow or ghosts cells)
If solitary pilomatricoma is very common, multiple pilomatricomas (usually up to 5) can be found in conjunction with a variety of abnormalities including myotonic dystrophy, Turner’s syndrome, trisomy 9, Gardner’s syndrome, Sotos syndrome, Rubinsten–Taybi syndrome, etc. [34–37].
Current studies have revealed that sporadic pilomatricomas have activation mutations in the cytoskeletal and cell-signaling protein β-catenin, encoded by CTNNB1 gene located on the 3p22–p21.3 region [38–42]. β-catenin is a 92-kDa protein involved in both WNT-signaling pathway and intercellular adhesion. It is suggested that WNT/β-catenin/LEF (lymphoid enhancer factor) pathway is activated in normal hair follicular matrix to induce differentiation toward hair shaft [43]. Activated WNT signaling leads to suppression of β-catenin phosphorylation and accumulated protein translocates to the nucleus, where it acts along with LEF to activate various transcription genes, such as MYC, CYC D1, etc. [44]. Nuclear accumulation of β-catenin can be demonstrated by using immunohistochemical studies and suggests either an activation of WNT-signaling pathway or a mutation that reduces β-catenin phosphorylation. Presence of β-catenin in the nucleus promotes cellular proliferation and was demonstrated in other tumors, such as colorectal carcinomas, melanoma, endometrial, pancreatic, and hepatocellular carcinomas [40, 45]. Mutations in CTNNB1 are located in the exon 3 region of the gene, which encodes the N-terminal phosphorylation sites. Missense mutations at these sites lead to β-catenin stabilization through decreased phosphorylation and degradation. In the skin, apart from pilomatricomas, mutations in exon 3 of CTNNB1 gene were also found in other tumors such as trichoepithelioma, basal cell carcinoma, and pilomatrical carcinoma, suggesting that β-catenin gene mutations contribute to these tumors tumorigenesis [40, 42, 45]. By immunohistochemistry, nuclear β-catenin, nuclear Lef-1, and cyclin D1 can be detected in the basaloid cells of pilomatricoma and pilomatrix carcinoma.
The role of bcl-2 in the histogenesis of pilomatricoma was recently reported [46]. Bcl-2 gene, located on chromosome 18, encodes an oncoprotein that blocks the apoptosis not only in various cells, such as lymphocytes, but also in the matrical follicular cells.
Fibrofolliculoma/Trichodiscoma
Clinical Features
Fibrofolliculoma and fibrodiscoma are follicular-derived benign cutaneous tumors, currently considered by many as part of the same histologic spectrum. They present as asymptomatic, small (1–4 mm), white-skin colored smooth papules. They are commonly found on the neck, upper chest, upper back, and face [47]. The lesions may range from one to several hundreds over a lifetime [48]. Fibrofolliculomas normally appear in the third and fourth decade of life (median age of 54 years) with no predilection for either sex [48]. The number and size of the fibrofolliculomas may increase with age; in cases with multiple lesions, they have the potential to be disfiguring, causing psychological burden [49].
Histopathology
Histopathologically, as the name suggests, fibrofolliculoma is a benign hamartomatous lesion with combined features of proliferation of hair follicle and perifollicular fibrous tissue. There are strands and cords of epithelial cells that are 2–4 cells thick, radiating from the infundibulum of a follicle that may be dilated and filled with keratinous material. The strands are surrounded by loose connective tissue containing fine collagen fibers with intervening mucin and partial or complete loss of elastic fibers. The epithelial strands may anastomose with each other or unite with the follicular infundibulum [50] (Fig. 4.3).
Fig. 4.3
Fibrofolliculoma—Histologically characterized by anastomosing strands and cords of epithelial cells radiating from the follicular infundibulum and surrounded by loose connective tissue with partial or complete loss of elastic fibers
On the other hand, trichodiscoma is a well circumscribed but nonencapsulated benign tumor with overlying flattened epidermis and folliculosebaceous units forming a collarette. It is composed of loosely arranged fascicles of fine collagen fibers intermingled with spindle-to-stellate-shaped fibroblasts in a background of mucinous stroma. There may be admixed prominent small blood vessels with PAS-positive basement membranes and few nerve fibers, predominantly at the periphery of the lesion [50].
Birt–Hogg–Dubé Syndrome (BHDS)
BHDS is a rare autosomal dominant inherited genodermatosis [48, 51]. BHDS is named after three authors (Canadian physicians Birt, Hogg, and Dube) who published a paper in 1977 describing family members with papular skin lesions on their face, forehead, scalp, and neck [50]. These three skin lesions characteristic of BHDS are fibrofolliculoma, trichodiscoma, and acrochordon. Acrochordon are very common lesions in general population and are rarely associated with BHDS [52, 53]. In one study, cutaneous lesions were found in 90 % (46/51) of families with BHDS [47]. For unknown reasons, Asian patients with BHDS are less likely to develop the cutaneous manifestations of the disease [54].
In addition to this skin lesion, people with BHDS are at increased risk of developing lung cysts, spontaneous pneumothorax, and kidney neoplasms [48]. Patients with fibrofolliculomas have a 50-fold increased risk for developing spontaneous pneumothorax, and a 7-fold increased risk for renal carcinoma [55]. It is important to recognize the possible association between fibrofolliculomas and BHDS as the diagnosis of the cutaneous lesion may lead to the early detection of life-threatening conditions, such as kidney neoplasms and pneumothorax. In one study, 72 % (18/25) families with kidney tumors were diagnosed after recruiting based on cutaneous manifestations of fibrofolliculoma [47].
Molecular Findings
In 2001, a BHD-associated gene locus was localized to chromosome 17p11.2 by linkage analysis [56]. Subsequently, truncating germline mutations were identified in a novel gene, the FLCN (BHD) gene (GenBank accession number AF517523), coding for a protein of unknown function [57]. The FLCN gene contains 14 exons and encodes folliculin, an evolutionary conserved protein of 579 aminoacids, with a central glutamic acid-rich coiled-coil domain, one N-glycosylation site, and three myristoylation sites, which has no major homology to any other human protein [58]. The function of folliculin is largely unknown. Somatic second-hit mutations in FLCN, identified in BHD-associated renal tumors, are consistent with a tumor suppressor function [59]. Loss of FLCN mRNA expression was found in renal tumors from patients with BHD, however, FLCN mRNA was reported to be strongly expressed in fibrofolliculomas. Loss of heterozygosity was not detected in fibrofolliculomas, suggesting that haploinsufficiency is sufficient to cause benign tumor growth in the skin and the mechanisms of tumorigenesis might differ in renal and skin tumors [60].
The mTOR (mammalian/mechanistic target of rapamycin complex 1) pathway, a key regulator of cell growth, proliferation, and metabolism, has been proposed to be involved in the pathogenesis of several hereditary hamartoma syndromes, including BHD. The precise role of FLCN in the mTOR pathway requires further elucidation, since several publications have reported contradictory effects of FLCN and it seems likely that FLCN has several functions. There are reports proposing that mTOR pathway is regulated by the interaction of FLCN and AMPK (5′AMP-activated protein kinase) mediated by FNIP1, a 130-kDa FLCN-interacting protein and FNIP2, an FNIP homologue [61, 62]. However, in a clinical trial, topical use of the mTOR inhibitor rapamycin was not an effective treatment for fibrofolliculomas [49]. FLCN may also be involved in the regulation of the TGF-beta signaling pathway, by deactivation of the transcription factor TFE3, and in overexpression of nuclear genes involved in the transcription and replication of the mitochondrial genome [63]. It has also been suggested that Drosophila BHD homolog (DBHD) may regulate maintenance of germline stem cells, downstream of, or in parallel with the JAK/STAT (which is necessary for germline stem cell self-renewal) and Dpp (a TGF-beta family member) signaling pathways [64]. Clarifying the roles of FLCN in the molecular pathogenesis of BHD-associated diseases might lead to the development of targeted therapies .
A germline FLCN mutation was found, by sequence analysis, in 84–88 % of families with BHD and 3–5 % of families had partial- or whole-gene deletions identified by other methods [47, 51]. Most of the reported pathogenic FLCN mutations are frameshift (52.9 %, 37/70) or nonsense mutations (10/70, 14.3 %) that lead to protein truncation, and followed by splice-site alterations (14/70, 20 %) [65]. The majority of mutations are deletions (45 %), substitutions (32–36 %), duplications (15 %), insertion/deletions (6 %), and insertions (2 %) [65, 66]. To date, the FCLN database includes more than 80 variants with > 53 unique germline mutations and > 30 SNPs. The unique germline BHD mutations have been reported in translated exons (4–14, excluding 8 and 10) of the BHD gene [47, 51]. The most frequent mutation in BHD patients is the insertion or deletion of a cytosine in the mononucleotide tract of eight cytosines (C8) in exon 11, predicted to cause a frameshift and prematurely truncate the mutant protein. This hot spot mutation occurs in about half of all BHD patients. Very few missense FLCN mutations were reported (e.g., 1523A→G [Lys508Arg]) [47, 51]. Mutations are located along the entire length of the coding region, with no genotype–phenotype correlations noted between type of mutation, location within the gene, and phenotypic disease manifestations [47].
The identification of FLCN defects in families with BHDS has led to a more accurate diagnosis. Formerly, BHDS was defined by the presence of multiple fibrofolliculomas (at least 5–10); the current proposed diagnostic criteria are based on clinical manifestations and the outcome of DNA testing [48, 56]. Patients should fulfill one major or two minor criteria of the following: Two major criteria are: (1) at least five fibrofolliculomas/trichodiscomas, at least one histologically confirmed, of adult onset and (2) pathogenic FLCN germline mutation. The three minor criteria are: (1) multiple lung cysts: bilateral basally located lung cysts with no other apparent cause, with or without spontaneous primary pneumothorax, (2) renal cancer, early onset (< 50 years) or multifocal or bilateral renal cancer, or renal cancer of mixed chromophobe and oncocytic histology, and (3) a first-degree relative with BHD [67].
Genetic Testing
FLCN is currently the only gene known to be associated with BHD. An MLPA (multiplex ligation-dependent probe amplification) kit for FLCN deletion and amplification analysis is available for genetic testing. The FLCN mutation database has been established by Wei and colleagues (http://www.skingenedatabase.com) and by the European BHD Consortium (Folliculin Sequence Variation Database). Mutation detection should be recommended for confirmation of diagnosis in suspected patients, as well as presymptomatic testing of at-risk relatives. Given the variable clinical manifestations of BHDS, genetic testing plays an important role in the identification of affected family members without skin tumors that could be at risk for developing renal tumors. The offspring of an individual with BHDS has a 50 % chance of inheriting the pathogenic variant (autosomal dominant) and prenatal diagnosis is possible if the FLCN pathogenic variant of an affected family member has been identified.
As per NCI recommendation, genetic testing for molecular diagnosis of a proband suspected of having BHDS should begin by sequence analysis of exon 11, as majority of the affected individuals have one of the two pathogenic variants found in exon 11. If a pathologic variant is not identified in exon 11, sequencing of the entire coding region of FLCN should be considered. If full-gene sequence analysis does not identify a pathogenic variant, deletion/duplication analysis of FLCN may be considered. Multigene panel by whole-exome sequencing may be considered in individuals with a clinical diagnosis of BHDS, but in whom no pathogenic variant in FLCN is identified. Surveillance in FLCN-mutation carriers and presymptomatic testing of family members of a confirmed BHD patient usually begins at the age of 18–20 years to allow counseling and informed consent before genetic testing, respectively. However, earlier testing and surveillance might be indicated in the families with a history of very early onset of pneumothorax or renal cancer [67].
Molecular Alterations in Cutaneous Adnexal Tumors with Sweat Gland Origin
Cylindroma/Spiradenoma
Clinical Features
Cylindromas and spiradenomas are difficult to distinguish clinically; they occur predominately in the head and neck region and present as multiple skin-colored small papules [68]. Spiradenoma can have a distinctive blue color and can also be painful [69]. The tumors appear early in adulthood and gradually increase in size and number throughout life, and may cause considerable disfigurement and discomfort .
Brooke–Spiegler syndrome (BSS) is an autosomal dominant disorder that predisposes affected patients to benign adnexal neoplasms including cylindromas, trichoepitheliomas, and/or spiradenomas. Ancell–Spiegler cylindromas (inherited disease of cylindromas) and Brooke–Fordyce trichoepitheliomas (inherited disease of trichoepitheliomas) were separate disorders first recognized in the mid-1800s [70–73]. With subsequent reports of the occurrence of trichoepitheliomas and cylindromas in the same patients, it became clear that these tumors were genetically related and the term BSS is now used for patients with multiple cylindromas, trichoeptheliomas, or a combination of both [69]. Some of the patients also develop spiradenomas, hence the triad of skin neoplasms (cylindromas, trichoepitheliomas, and/or spiradenomas) became characteristic of BSS [74].
Any or all of these tumors may develop at any point in life and there is variable penetrance within affected families [75]. Malignant transformation of these tumors is rare, but some studies suggest that tumors occurring in the setting of BSS can be more aggressive than their sporadic counterparts [76, 77]. Although BSS is inherited in autosomal dominant pattern, there is reduced penetrance in males with cylindromas and trichoepitheliomas occurring more commonly in females [78].
Histological Features
Cylindromas and spiradenomas are considered as being part of the same histologic spectrum and are tumors with eccrine or apocrine differentiation [79]. Trichoepitheliomas are follicular-derived lesions.
Cylindromas are histologically characterized by a dermal nodule composed of irregularly arranged islands of basaloid cells, sometimes admixed with small duct-like structures, surrounded by a thin eosinophilic band of hyaline material, arranged in a characteristic “jigsaw puzzle” pattern [80]. Most of the tumor islands show two cell types: a peripheral cell with a palisading dark-staining nucleus and a more centrally located larger cell with a vesicular nucleus. Both cell types are embedded in a stroma with loose collagen containing an increased number of fibroblasts . (Fig. 4.4)
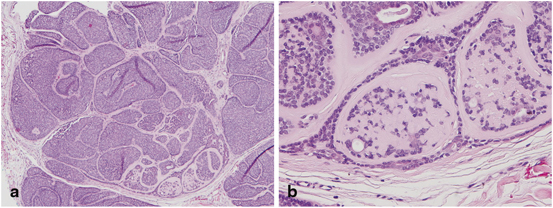
Fig. 4.4
Cylindroma/spiradenoma—Histologically characterized by a dermal nodule composed of “jigsaw puzzle”—arranged islands of basaloid cells with small duct-like structures, surrounded by an eosinophilic hyaline material (a). There are two cell types: a peripheral cell with a palisading dark-staining nucleus, and a more centrally located larger cell with a vesicular nucleus (b)
Trichoepitheliomas are dermal tumors showing focal continuity with the epidermis in up to one third of cases. They are composed of uniform basaloid cells with peripheral palisades, arranged in variably sized nests or with a cribriform pattern, surrounded by dense stroma and fibroblasts [81]. Epithelial structures resembling hair papillae or abortive hair follicles (papillary mesenchymal bodies) and small keratinous cysts lined by stratified squamous epithelium may be seen. The desmoplastic trichoepithelioma variant is histologically characterized by a dense and hypocellular stroma, with fewer elastic fibers and more acid mucopolysaccharides.
Spiradenomas are dermal tumor nodules formed by nests of two types of epithelial cells arranged in cords, with tubular or alveolar differentiation, embedded in a background of edematous and vascular stroma [82]. The two cell types comprise small dark basaloid cells with hyperchromatic nuclei and a more frequent larger cell with a pale nucleus (usually located in the center of the clusters). The cells are PAS negative, but droplets of PAS-positive hyaline material may be present in some areas. Duct-like structures, infiltrating lymphocytes, and irregular, thin fibrous bands containing blood vessels are often present within the tumor lobules .
Brooke–Spiegler Syndrome
Linkage analysis in families with multiple cylindromas first mapped the susceptibility gene to a single locus on chromosome 16q12–q13 [83, 84]. The CYLD gene was identified by positional cloning and germline mutations subsequently found in affected families [85]. The CYLD gene consists of 20 exons, of which the first three are untranslated (GenBank NM-015247). It encodes a 956 amino acid protein with a molecular weight of approximately 120 kDa. The mutations are scattered throughout the midportion and C-terminal region of the CYLD gene with 60 % clustering in the C-terminal region (exons 16–20), but not limited to one particular domain. The CYLD gene encodes the cylindromatosis protein (CYLD) (GenBank NP_056062), also known as ubiquitin-specific-processing protease CYLD, and ubiquitin carboxyl-terminal hydrolase CYLD. CYLD has moderate homology with proteins of the ubiquitin-specific protease class and it has enzymatic deubiquitinase activity. CYLD regulates cell functions including inflammation and cell proliferation through removal of ubiquitin of target proteins [86].
A total of 51 distinct germline CYLD mutations have been reported in the literature. The majority of the mutations are frameshift (41 %, 21/51) and nonsense (35 %, 18/51), followed by missense (14 %, 7/51), and putative splice site (10 %, 5/51) [86]. Eighty-six percent (44/51) of the mutations are predicted to result in truncated proteins. The most common reported mutation in CYLD is c.2806C > T, followed by c.2272C > T, c.2305delA, c.2172delA, and c.1112C > A. The overall CYLD mutation detection rate in BSS is 84 % (73/87) [86]. No correlation has been detected between CYLD mutations and a specific phenotype [86].
CYLD function has been reported primarily as a negative regulator of NF-κB signaling, a pathway shown to stimulate cell proliferation in a variety of tissues [87, 88] (Fig. 4.5). The NF-κB pathway is induced by a wide variety of stimuli including tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1). NF-κB suppression results in severe defects in the development of epidermal appendages, including hair follicles and sweat glands. However, the mechanism of aberrant NF-κB signaling and oncogenesis in the skin remains to be established. CYLD regulates the NF-κB pathway by deubiquitination of various targets; TRAF2 and its associated proteins; RIP1, TAK1, TRAF7, TRAF6 (occurs through a signal adaptor protein p62), and NEMO (NF-κB essential modulator). CYLD-mediated deubiquitination of target proteins subsequently inhibits downstream dissociation of IKK from NF-κB, leading to inhibition of the NF-κB pathway [89–95]. Loss of CYLD activity removes the inhibition of the NF-κB pathway, ultimately stimulating cell proliferation .
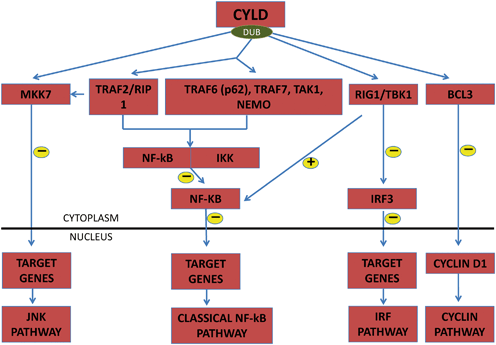
Fig. 4.5
Simplified schematic showing possible mechanisms of CYLD regulation of different signaling pathways of cell survival and proliferation through deubiquitination (DUB). Arrows denote multistep or incomplete pathways. CYLD deubiquitinates TRAF and its associated proteins RIP1, TRAF6 through the adaptor protein p62, TRAF7, TAK1, and NEMO, which inhibit downstream dissociation of NF-κB and IKK. CYLD-mediated deubiquitination of MKK7 and BCL-3 deactivates downstream JNK and cyclin D1 pathway, respectively. CYLD also negatively regulates IRF3 pathway by deubiquitination of RIG1 and TBK1
CYLD is also a negative regulator of JNK pathway, which plays a role in apoptosis, cell survival, and proliferation [96]. This regulation occurs in response to stimulation by several cytokines. Deubiquitination of TRAF2 by CYLD inhibits MKK7 activation and downstream JNK signaling [97, 98]. Also, CYLD deubiquitinates RIG-1/TBK1, which inhibits downstream activation of the growth promoting IRF3 signaling pathway [99, 100].
BCL-3 is a transcription factor that stimulates expression of cyclin D1. CYLD-mediated deubiquitination of BCL-3 prevents its translocation from the cytoplasm to the nucleus in response to UV light and subsequently inhibits the cyclin pathway, a cell cycle regulatory pathway. CYLD also affects cell proliferation and cycling by targeting Plk1 and histone deacetylase-6 (HDAC6) [101, 102]. It also has been shown to modulate cell migration via microtubule assembly and ion channel activity by deubiquitination of TRPA1 [103, 104].
The functions of CYLD still need further investigation, but there are multiple reports proposing CYLD as a tumor suppressor gene. Truncating mutations are found to be the most common tumor-predisposing germline mutations in CYLD (approximately 90 %). Most of the tumors show loss of heterozygosity (LOH) of the wild-type allele at the CYLD locus (chromosome 16q12–13) and some tumors without LOH have somatic mutations of CYLD [83, 85, 105–107]
Genetic Testing
CYLD mutation detection rate in BSS is relatively high, with 84 % of affected patients having mutations in the gene. Genetic testing for CYLD mutations, exons 16–20 in particular, is useful for identifying affected individuals as over 75 % of BSS families have a mutation in these regions [86]. In families with history of a severe phenotype and a confirmed CYLD mutation in a parent or sibling, prenatal testing for pregnancies should be performed [86]. Early identification of mutations and management of skin appendageal tumors can potentially minimize psychological burden due to disfigurement and promote early detection of malignant lesions. Further studies to identify positive and negative regulators of CYLD are necessary for a complete understanding of the mechanisms by which CYLD regulates tumorigenesis, as well as for the development of novel therapies for adnexal tumors .
Hidradenoma
Recent studies have shown that the t(11;19)(q21;p13) translocation seen in certain salivary gland tumors (such as mucoepidermoid carcinoma and Warthin’s tumor) resulting in fusion of N-terminal CREB-binding domain of TORC1 to the Notch coactivator MAML2 was also identified in clear cell nodular hidradenoma. By RT-PCR analysis, TORC1–MAML2 fusion transcript was revealed, consisting of exon 1 of TORC1 fused to exons 2–5 of MAML2 [108].
Molecular Alterations in Cutaneous Adnexal Tumors with Sebaceous Origin
Clinical and Histologic Features of Sebaceous Neoplasms
There is a wide spectrum of sebaceous neoplasms ranging from hamartomatous to benign to malignant entities and their classification is often controversial. The hamartomatous, ectopic, and some benign sebaceous lesions, such as sebaceous hyperplasia, are almost exclusively sporadic and there is currently no description of their association with systemic syndromes. Therefore, considering the scope of our publication, we further describe sebaceous lesions that may potentially have this association .
Sebaceous adenomas are benign tumors derived from sebaceous glands that occur commonly on the head and neck region of older individuals as tan-yellow, small (less than 5 mm) papules. Histologically, they have a lobular, organoid growth pattern, are well-circumscribed, and often connected to epithelial surface. Sebaceous adenomas have an increased number of basaloid cells within the lobules: more than the normal two-cell layers but less than 50 % of the entire lesion [109] (Fig. 4.6).
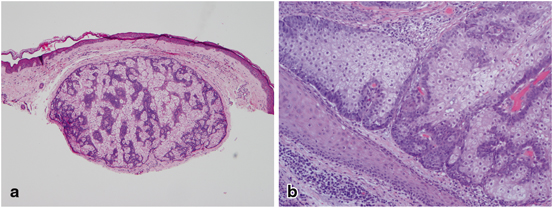
Fig. 4.6
Sebaceous adenoma—Histologically there is a well-circumscribed lobular, organoid growth pattern with an increased number of basaloid cells within the lobules (a), more than the normal two-cell layers but less than 50 % of the entire lesion (b)
Sebaceomas (sebaceous epitheliomas) are usually larger lesions, typically ranging from 5 to 10 mm, are fleshy yellow and also occur in the head and neck region. Histologically, the sebaceomas have a lobular growth pattern, similar to sebaceous adenomas, but differ from them by the marked increased number of basaloid, germinative cells (more than 50 % of the lesion), and lack the organoid pattern. Often sebaceomas involve dermis, but sometimes, connection with epidermal surface is noted. They are well-circumscribed lesions and there is no significant cytologic atypia or increased number of mitotic figures [110]. However, due to its higher proportion of basaloid, germinative cells and sometimes less obvious presence of mature sebocytes, sebaceomas may be difficult to distinguish from basal cell carcinomas. The lack of peripheral retraction artifact and associated myxoid stoma is helpful in the differential diagnosis .
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


