Fig. 5.1
Tumor Angiogenesis. Blood vessels, tumor cells, and the cells of the tumor microenvironment are schematically depicted. Enlarged image (right panel) shows the cross section of a blood vessel.
Mechanisms of Tumor Angiogenesis in Melanoma
There are several modes of tumor vascularization. For many years, the process of tumor vascularization was thought to mainly involve sprouting of new blood vessels from pre-existing vasculature (sprouting angiogenesis). However, in recent years, additional mechanisms have been recognized, such as vascular co-option, intussusceptive angiogenesis, mosaic vessels, bone marrow-derived vasculogenesis, and vasculogenic mimicry [12–15].
Sprouting Angiogenesis – the growth of new capillary vessels from pre-existing ones – has been the most studied mechanism of neo-vascularization over the years [11, 12, 16] (Fig. 5.2). It involves endothelial cell proliferation, migration, and tube (vascular channel) formation. There are three sequential steps; quiescence, activation, and resolution. Every step involves well-coordinated molecular signaling. Mechanistically, it begins with basement membrane degradation of the endothelium and disruption of this monolayer [12]. When a quiescent vessel senses an angiogenic signal (e.g. growth factors such as VEGF, ANG-2, FGFs, and cytokines) released by the tumor cell, pericytes first detach from the vessel wall and liberate themselves from the basement membrane by proteolytic degradation (mediated by matrix metalloproteinases). Endothelial cells loosen their junctions, the nascent vessel dilates, plasma proteins and others extravasate to form an angio-competent milieu. To form a perfused tube (a new vessel) one endothelial cell, known as the ‘tip cell’, becomes selected to lead the tip. VEGFRs, neuropilins, and the NOTCH receptors are critical for this step. The neighbors of the tip cell, known as the ‘stalk cells’, divide and establish the lumen followed by initiation of blood flow, deposition of basement membrane, and pericyte coverage. During maturation, stalk cells transform into phalanx cells. The end result of this process is a new perfused vascular channel from a pre-existing one.
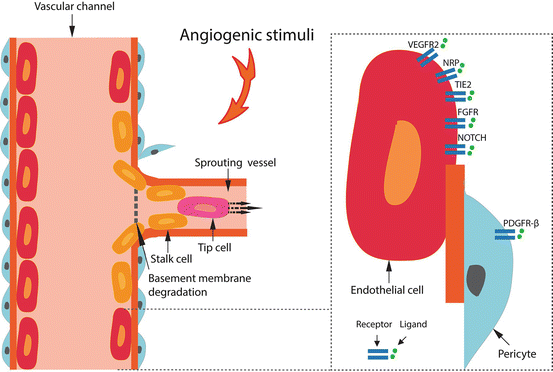
Fig. 5.2
Sprouting Angiogenesis. The process of neo-vascularization in sprouting angiogenesis is shown. Enlarged image (right panel) shows the key ligand-receptor interactions on the surface of the endothelial cells.
Tumor Microenvironment During Angiogenesis
Tumor angiogenesis was first thought to be fueled by neoplastic cells, however emerging evidence suggests that the tumor microenvironment and the immune cell subsets are as important for regulating this process (Table 5.1) [17, 18]. There appears to be a complex but well orchestrated interplay between the neoplastic cells, immune cells, and the vascular endothelium [12, 19]. Tumor cells disrupt normal tissue homeostasis by altering its gene expression to secrete molecules (e.g. growth factors and cytokines) and cellular components to recruit other cell types. Early in the course of tumor growth, hypoxia leads to activation of hypoxia-inducible factor that follows a rapid increase in blood vessel formation. Cells of the tumor microenvironment are heterogeneous in origin, and can be derived from the stroma, blood vessels, or the bone marrow [16].
Table 5.1
Tumor microenvironment in angiogenesis
Tumor microenvironment | Major constituents |
|---|---|
1. Cells (a) Tumor cells (b) Immune cells (c) Pericytes | Melanoma cells Monocytes, macrophages, myeloid derived suppressor cells, dendritic cells, natural killer cells, neutrophils, mast cells, eosinophils, T and B cells, plasma cells |
2. Stroma (a) Extracellular matrix (b) Fibroblasts (c) Myofibroblasts (d) Endothelial cells | Collagen, fibronectin, laminin, proteoglycan |
3. Soluble factors (a) Pro-angiogenic (b) Anti-angiogenic (c) Proteases | VEGFs, PDGFs, Ang-1/2, PROK-1/2, FGFs, EGF, IGF, HGF, PIGF Thrombospodin-1, angiostatin, endostatin Matrix metalloproteinases, u-PA, elastase |
Angiogenic Factors in Melanoma
A number of pro-angiogenic growth factors essential for melanoma growth and metastasis have been characterized. They function via the classic ligand-receptor interaction; the growth factor released from the tumor cell or other cells within the microenvironment binds to its specific receptor expressed on the endothelial cell and enhances signaling leading to endothelial cell proliferation and migration (Fig. 5.2). While these growth factors are typically secreted or released by the tumor cell, some are let-off by the stromal cells, bone marrow derived progenitor cells, and/or inflammatory cells. Some of the key pro-angiogenic growth factors, their receptors, and contribution to the angiogenesis program in melanoma are reviewed.
The VEGF (Vascular Endothelial Growth Factor) Family
VEGF family is a large group of growth factors including VEGF-A, -B, -C, -D, -E, and placental growth factor-1 and -2 (PIGF-1 and -2) [20–22]. VEGF-A (also referred as VEGF) is the main component that regulates angiogenesis in health and disease [23]. VEGFR-2 is the predominant receptor tyrosine kinase that mediates VEGF signaling in endothelial cells and that drives VEGF-mediated angiogenesis. VEGFs bind to three structurally similar tyrosine kinase receptors VEGFR-1 (Flt-1), VEGFR-2 (Flk-1, KDR) and VEGFR-3 (Flt-4) to stimulate blood vessel proliferation and angiogenesis [21]. VEGFR-1 and -2 are expressed almost exclusively on endothelial cells, while VEGFR-3 is necessary for blood vasculature during early embryogenesis, but later becomes a key regulator of lymphangiogenesis, the formation of new lymphatic vessels. While VEGFR-1 and -2 are involved in angiogenesis by VEGF-A isoform, VEGFR-3 is involved in lymphangiogenesis by VEGF-C and -D. Neuropilin-1 and -2 are members of the VEGF family that function as co-receptors to enhance binding to VEGFR-2, but also can signal independently.
VEGF is upregulated in melanoma, but is not expressed in normal melanocytes [24]. Increased expression of VEGF and VEGFRs in primary cutaneous melanoma as well as increased microvascular density strongly correlates with disease progression [25–27]. Serum VEGF levels are increased in melanoma patients compared with healthy controls [28].
The PDGF (Platelet-derived Growth Factor) Family
PDGF is a family of five growth factors (PDGF-AA, -AB, -BB, -CC, and -DD) that exert their biological effects through tyrosine kinase receptors, PDGFR-α and -β [21, 22]. The PDGF isoforms differ in their receptor specificity. The -A and -C chains bind only to PDGFR-α, and the -D chain binds only to PDGFR-β, whereas the -B chain binds to both. PDGFs together with angiopoietins and TGF-β have critical roles in endothelial cell-pericyte interaction [29]. During angiogenesis, endothelial cells produce PDGF-BB that stimulates PDGFR-β-expressing pericytes, and result in proliferation and migration of pericytes. Pericytes provide coverage of the vessel wall to stabilize the channels. Either PDGFR inhibition or pericyte deficiency leads to vessel leakage, tortuosity, and immature vessel formation. In the context of tumor angiogenesis, pericyte coverage of the vessel walls appears to be protective against metastasis as endothelial channels lined with pericytes limit tumor cell intravasation as opposed to those that are loosely assembled and leaky [30].
Melanomas show enhanced expression of PDGF-AA, −BB, and PDGFR-α [31]. In highly metastatic melanoma cell lines, expression of PDGFR-α is significantly elevated as compared to PDGFR-β.
The FGF (Fibroblast Growth Factor) Superfamily
FGFs activate receptors (FGFR1-4) on endothelial cells or indirectly stimulate angiogenesis by inducing the release of angiogenic factors from other cell types [12]. Low levels of FGF are required for the maintenance of vascular integrity. Aberrant FGF signaling promotes tumor angiogenesis.
bFGF (basic FGF or FGF-2) has been well studied in melanocytic tumors. It is expressed in melanoma cells but not in normal melanocytes [32]. Targeting bFGF in melanoma cells decrease tumor growth in vitro and in vivo [33].
ANG-TIE Signaling
The ANG (Angiopoietin) family is composed of at least three ligands (ANG-1, -2, and -4) and two tyrosine kinase receptors, TIE-1 and TIE-2. ANG-1 functions as a TIE-2 agonist, stimulates mural coverage and vessel tightness, and is critical for maintenance of endothelial cell quiescence. Whereas ANG-2 when stimulated by angiogenic factors antagonizes the ANG-1 and TIE-2 signaling to enhance mural cell detachment, vascular permeability, and endothelial cell sprouting. Thus, ANG-1/TIE-2 and ANG-2 function in a reciprocal manner as anti- or pro-angiogenic, respectively.
Elevated serum levels of ANG-2 are reported in patients with melanoma compared with healthy individuals [34]. Circulating levels of ANG-2 correlates with stage of disease and overall survival.
NOTCH Signaling
There are three ligands (Delta-like 4, Jagged-1 and -2) and four NOTCH receptors (NOTCH 1–4) [35]. DLL4 and NOTCH signaling is critical for generating perfused vessels, tip cell selection, stalk cell proliferation [35]. The activity of this signaling is low during vessel quiescence. In tumor angiogenesis, inhibition of Delta-like 4/NOTCH signaling induces more but hypoperfused vessels, resulting in growth inhibition.
WNT Signaling
Endothelial cells express various types of WNT ligand and their frizzled (FZD) receptors, of which several stimulate endothelial cell proliferation [36]. NOTCH and WNT activate each other in a reciprocal-feedback system [36]. WNT and NOTCH result in a similar phenotype in endothelial cells, characterized by branching defects, loss of venous identity and aberrant vascular modeling.
Angiogenesis in Melanoma: The Therapeutic Implications
Targeting VEGF
The concept of anti-angiogenesis therapy was coined by Folkman and colleagues in 1971 based on the observation that tumors do not grow beyond a minimal size of 1–2 mm3 without new vessel formation and therefore inhibiting angiogenesis could prevent tumor progression [37]. VEGF was discovered as the primary mediator of tumor angiogenesis soon followed by generation of monoclonal antibody against this growth factor and FDA approval in 2004 [38]. A plethora of pre-clinical and clinical studies on VEGF and its inhibition including in melanoma concluded its lack of effectiveness as a single agent [38]. However, combining it with other drugs have shown clinical benefit, and thus it is approved and used only in combination with other drugs in several solid tumors [12]. Many patients are refractory or acquire resistance to VEGF inhibitors, and biomarkers to identify responders are lacking [39].
In melanoma, its combinatorial partner(s) are vigorously being sought, immune checkpoint blockade (e.g. anti-CTLA-4, anti-PD1, anti-PDL1) currently receiving immediate attention. It is likely that in the near future key drivers of angiogenesis beyond VEGF will be identified as well as bypass mechanisms when a major endothelial growth factor such as VEGF is inhibited along with a search for immune modulating and other combinatorial agents that act in synergy, thus leading to effective angiogenesis-based therapeutics. Readers can view the ongoing clinical trials targeting VEGF or others in melanoma via www.clinicaltrials.gov.
Bevacizumab (Table 5.2)
This is a recombinant humanized monoclonal antibody that neutralizes VEGF isoforms [40]. Although there are many drugs that inhibit VEGF and its signaling (e.g. sorafenib, sunitinib), bevacizumab is the most commonly studied and used agent to date and considered as a prototype for anti-angiogenesis based treatments due to its selectivity. Bevacizumab combined with other neoplastic agents (e.g. interferon-alpha, chemotherapy) has been approved for the treatment of advanced non-small cell lung cancer, breast cancer, colorectal cancer, glioblastoma multiforme, and renal cell carcinoma [12]. It is also licensed for the intravitreal treatment of choroidal neovascularization secondary to age-related macular degeneration [41].
Table 5.2
Bevacizumab in the treatment of cancer
Bevacizumab as single agent | Ineffective |
|---|---|
Bevacizumab in combination [interferon or chemotherapy] | Approveda for the treatment of Metastatic colorectal cancer Metastatic non-small cell lung cancer Metastatic renal cell cancer Recurrent glioblastoma multiforme |
Clinical trials in melanoma concluded that Bevacizumab has little activity as a single agent for the treatment of advanced melanoma [42]. However combining VEGF inhibitors with other agents may be of interest. In some solid cancers, even though bevacizumab as monotherapy was ineffective combining with interferon or chemotherapy, lead to marked clinical improvement and is currently approved for the treatment of these tumors (e.g. colorectal cancer, renal cell carcinoma, non-small cell lung cancer, breast cancer, ovarian cancer, and glioblastoma multiforme) [43].
This drug has been found to initially reduce tumor perfusion, vascular volume, and microvascular density, findings referred to as ‘vascular normalization’ [44]. This phenomenon has been proposed as a plausible explanation for the improved activity of radiotherapy and cytotoxic agents given in combination with bevacizumab, as tumor normalization improves tissue oxygen levels and drug delivery. However, unlike other cancers, the search for interferon or a chemotherapeutic agent in combination with bevacizumab with improved activity for melanoma has not yet been successful. It has been tested in combination with low dose interferon alpha-2a (plus dacarbazine) [45], high-dose interferon alpha-2b [46], or several chemotherapeutic agents in metastatic melanoma [42], but did not show significant clinical responses. Adverse effects included neutropenia, peripheral neuropathy, arterial thromboembolic events and hypertension [42].
One of the largest initial trials was the BEAM study with 214 patients investigating bevacizumab (in combination with carboplatin and paclitaxel). In this randomized phase 2 study there was no improvement in progression-free survival. At 17-month follow-up, bevacizumab prolonged the median overall survival (12.3 versus 8.6 months), with a non-significant 21% reduction in the hazard of death [42]. Bevacizumab and fotemustine showed an overall survival of 20 months in 20 patients [47]. A phase II trial combining bevacizumab and high-dose interferon alpha-2b in advanced melanoma that accrued 25 patients showed a median overall survival of 17 months [46]. In a phase II trial combining bevacizumab and temsirolimus (CTEP 7190/Mel47) for unresectable stage III/IV melanoma, three patients showed partial response (17.7%), 9 patients stable disease at 8 weeks, and 4 patients had progressive disease [48]. Temozolomide and bevacizumab in 62 patients revealed overall survival of 9.6 months (SAKK 50/07) [49]. In another phase II trial (N0775), temozolomide and bevacizumab or nab-paclitaxel, carboplatin and bevacizumab in patients with unresectable stage IV melanoma were evaluated in 93 patients and showed median overall survival of 12.3 months and 13.9 months, respectively [50].
In preclinical studies VEGF has been associated with anti-tumor responses, including suppression of dendritic-cell maturation, proliferation of regulatory T cells, inhibition of T-cell responses, and accumulation of myeloid-derived suppressor cells [51–55]. Thus, combining VEGF inhibition with immune checkpoint blockers (e.g. anti-CTLA-4) plus given the success of ipilimumab in the clinical setting was the next logical step. In a recent study bevacizumab and ipilimumab combination resulted in partial responses in 8 patients and stable disease 22 patients in a phase I trial of 46 patients [56]. Disease control rate in this study was 67.4% and median survival was 25.1 months. In an earlier study, ipilimumab, when compared with gp100 peptide vaccine improved the median overall survival from 6.4 to 10.1 months [3]. Therefore, the study combining ipilimumab with bevacizumab suggests worthy of future pursuit towards this combination. Notably, inflammatory toxicities were generally higher with the combination therapy than ipilimumab alone but were manageable [56].
Bevacizumab in the Adjuvant Setting
There is a need to improve adjuvant treatment for patients who have melanoma and are at high risk of disease recurrence after surgery. Adjuvant use of bevacizumab in this context was examined (AVAST-M study). A large phase III open-label multicenter clinical trial published in 2014 randomized 1343 patients with resected cutaneous melanoma at high risk for recurrence (stage IIB, IIC, and III) to either bevacizumab group (n = 671) or the observation group (n = 672). One hundred thirty four (96%) patients in the bevacizumab died of melanoma versus 139 (95%) in the observation group. There was no significance in overall survival between the groups at a median of 51 weeks of treatment. At this point, one can conclude that adjuvant use of bevacizumab in melanoma is not beneficial. This finding is not surprising, as the adjuvant use of this drug has not improved survival in patients with any other tumor type to date [57]. Nevertheless, longer follow up to meet the 5-year survival end point will determine the final beneficial effects and risks of this drug in melanoma [58].
Bevacizumab in Uveal Melanoma
Uveal melanoma is a subtype of melanoma that arises from melanocytes of the uveal tract [59]. Apart from its difference from cutaneous melanoma in its anatomic location, developmental pathways, clinical characteristics, genetic aberrations, and prognosis, it metastasizes through the hematogenous route, typically to the liver, and is an angiogenic tumor [59]. Levels of VEGF are significantly elevated in patients with metastatic disease compared to patients without metastases [60]. IGF-1 has been shown to stimulate secretion of VEGF in retinal pigment epithelial cells and IGF-1 signaling may also stimulate tumor angiogenesis in uveal melanoma liver metastases [61].
Related posts:
 Infantile Hemangioma: New Insights on Pathogenesis and Beta Blockers Mechanisms of Action
Infantile Hemangioma: New Insights on Pathogenesis and Beta Blockers Mechanisms of Action
 Angiogenesis: General Concepts
Angiogenesis: General Concepts
 Chemoprevention and Angiogenesis
Chemoprevention and Angiogenesis
 The Role of Angiogenesis in the Development of Psoriasis
The Role of Angiogenesis in the Development of Psoriasis
 Angiogenesis and Pathogenesis of Port Wine Stain and Infantile Hemangiomas
Angiogenesis and Pathogenesis of Port Wine Stain and Infantile Hemangiomas
 Potential Role of Angiogenesis and Lymphangiogenesis in Atopic Dermatitis: Evidence from Human Studies and Lessons from an Animal Model of Human Disease
Potential Role of Angiogenesis and Lymphangiogenesis in Atopic Dermatitis: Evidence from Human Studies and Lessons from an Animal Model of Human Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


