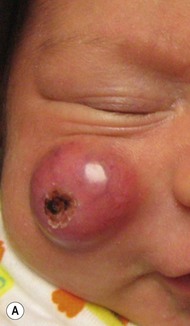
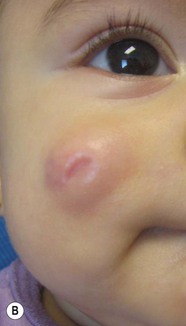
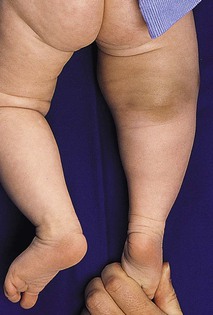
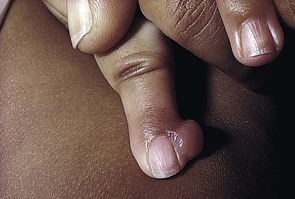
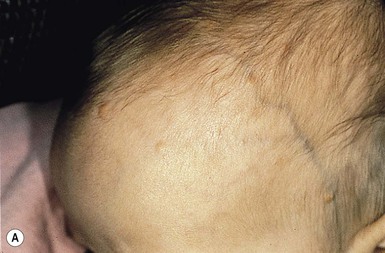
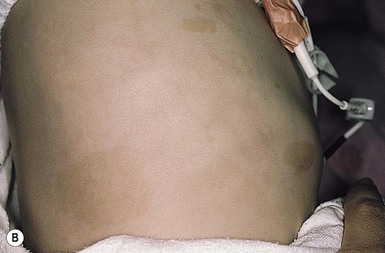
Julie S. Prendiville A wide variety of conditions affecting the skin and subcutaneous tissues present as papulonodular lesions, or ‘lumps and bumps.’ Benign and malignant neoplasms, hamartomas, and inflammatory and infectious disorders, as well as a number of infiltrative diseases, can be included in this category. Some of these conditions are discussed in detail in other chapters. This section deals with a group of nonmalignant disorders that present as discrete, circumscribed skin lesions or hamartomas in newborns and young infants. The fibromatoses represent a diverse collection of mesenchymal tumors characterized by fibroblastic–myofibroblastic proliferation.1 They are locally invasive neoplasms that do not metastasize but which may recur following surgical excision. They vary in clinical behavior, from benign lesions that regress spontaneously to aggressive life-threatening tumors. They can be solitary or multifocal, and may exhibit skin, soft tissue, bone, or visceral involvement. Most of these tumors are sporadic, but some occur in a familial setting. The fibromatoses are classified as juvenile or adult (Box 26.1).1 The juvenile fibromatoses are a unique group of fibroblastic–myofibroblastic proliferations that present at birth or in the first years of life (Table 26.1), accounting for approximately 12% of pediatric soft tissue tumors.1 Adult-type fibromatoses are occasionally observed in infancy and childhood.1 The fibromatoses have also been subdivided according to the site of fibrous tissue overgrowth into superficial or fascial, and deep or musculoaponeurotic (desmoid type).2 The term infantile myofibromatosis was introduced by Chung and Enzinger3 in 1981 to designate a disorder previously described under numerous synonyms, including congenital multiple fibromatosis, diffuse congenital fibromatosis, multiple congenital mesenchymal tumors, and multiple vascular leiomyomas of the newborn. There are three clinical patterns of presentation: solitary infantile myofibroma; multicentric infantile myofibromatosis, with multiple lesions in the skin, soft tissues, and bone; and generalized infantile myofibromatosis, in which there is also visceral involvement.1 Over 80% of myofibromas present in the first 2 years of life, and 60% are apparent at birth or shortly thereafter.3,4 Lesions may be superficial or deep, involving the skin, subcutaneous tissues, and muscle. They appear clinically as discrete, rubbery, firm to hard nodules measuring from 0.5 to 8 cm in diameter (Fig. 26.1). Cutaneous myofibromas may be skin-colored or have a prominent vascular appearance, resembling hemangioma. Sites of predilection for solitary lesions are the head, neck, trunk, and upper extremities. In the multicentric and generalized forms there are multiple and widespread myofibromas, numbering from a few to over 100 (Fig. 26.2).5 Skin and soft tissue lesions are asymptomatic and usually cause little morbidity. Rarely, a myofibroma presents with surface ulceration or an atrophic morphology (Fig. 26.3).4,6 Joint contractures have been observed with extensive limb lesions.7 In the multicentric form of the disease, myofibromas in the skin and soft tissues are associated with multiple lytic bone lesions. These may be extensive and can involve any bone.5 Progression in size and number has been observed during infancy.5 The bone tumors eventually stabilize, and spontaneous healing occurs with complete regression during the first few years of life. The development of sclerotic borders around lytic areas may be an early sign of regression.5 In most cases, there are no clinical signs or symptoms of bone disease. Pathologic fractures may occur rarely and usually heal without residual deformity.5,8 Vertebral body collapse has been described, with residual loss of vertebral height in early childhood.5 There are reports of spinal cord compression resulting from solitary or multicentric lesions involving the spinal canal.9 Solitary infantile myofibroma may also occur in the orbit.10 The much rarer generalized form of infantile myofibromatosis is characterized by involvement of visceral organs in addition to skin, soft tissue, and bone tumors. The gastrointestinal tract, heart, and lungs are most frequently affected. Involvement of the central nervous system is rare. Myofibromatosis in visceral organs is locally invasive and may severely compromise organ function. Cardiopulmonary, gastrointestinal, and hepatobiliary complications can be fatal, particularly in the newborn period or early infancy.11 There have been two reports of associated aneurismal dilatations of arteries as observed in fibromuscular dysplasia.12 Multiple skin and soft tissue tumors may occasionally occur in the absence of bone or visceral involvement.1 Conversely, bone involvement has been observed in association with a single soft tissue lesion,11 or in the absence of skin lesions.5 The pathogenesis is unknown. Most cases are sporadic. Familial occurrence is associated with autosomal dominant inheritance.13 Myofibromatosis may be suspected by the presence of firm, cutaneous and subcutaneous nodules. A biopsy is required to confirm the diagnosis. All three forms of infantile myofibromatosis show interlacing fascicles of spindle-shaped fibroblasts.1 Central vascular areas resembling hemangiopericytoma are variably present. Focal necrosis, calcification, hyalinization, macrophages containing hemosiderin, and chronic inflammation may be seen.1 A giant cell variant containing multiple multinucleated giant cells has also been described. There is positive immunoreactivity for vimentin and actin, consistent with the presumed myofibroblastic derivation of the tumor; desmin staining is variable. Electron microscopy shows cells with features of both fibroblasts and smooth muscle cells. Infants with cutaneous myofibromas should be evaluated for bone and visceral involvement, particularly when there are multiple lesions. Recommended initial investigations include a skeletal survey, chest X-ray, echocardiogram, and abdominal imaging studies.11 Infantile myofibromatosis can be distinguished from other pediatric soft tissue tumors by histopathologic examination of biopsy material. These include other forms of fibromatosis, as well as congenital fibrosarcoma, leiomyoma and leiomyosarcoma, neurofibroma, metastatic neuroblastoma, hemangioma, hemangiopericytoma, chondromatosis, stiff skin syndrome, and nodular fasciitis.1 The prognosis for infantile myofibromatosis is good in the absence of visceral involvement. Lesions in the skin and soft tissues show spontaneous involution during the first few years of life, sometimes leaving residual areas of skin atrophy or hyperpigmentation (Fig. 26.4). Bone lesions also regress spontaneously, usually without significant disability or residual radiologic change. They do not interfere with enchondral bone growth.5 The prognosis is grave for newborns with visceral disease, in whom a mortality rate of 76% has been documented.11 Surgical excision may be necessary to obtain tissue for diagnosis. Otherwise, excision should be limited to lesions that result in functional impairment or severe cosmetic disability.7,11 The role of chemotherapy or radiation for symptomatic, recurrent, or nonresectable disease is not established. Successful treatment of life-threatening generalized infantile myofibromatosis and multicentric disease using low-dose chemotherapy has been reported.14 Although desmoid fibromatosis has traditionally been considered a deep fibromatosis of adulthood, with abdominal, intra-abdominal, and extra-abdominal variants, a specific juvenile subset is recognized.1 Description of this entity under a variety of synonyms, including among others aggressive fibromatosis of infancy, musculoaponeurotic fibromatosis, desmoma, and fibrosarcoma grade 1 desmoid type, has led to confusion in the literature.15 Up to 30% of juvenile desmoid tumors present in the first year of life, and congenital cases have been reported.1,16 Infantile desmoid-like or aggressive fibromatosis involves deep tissues and is generally extra-abdominal.1 The usual clinical presentation is a slowly growing, nontender subcutaneous mass that has been present for weeks or months (Fig. 26.5). Sites of predilection in children are the head and neck, extremities, shoulder girdle, trunk, and hip regions. The abdomen, retroperitoneum, spermatic cord, and breast may also be involved. Rarely, there are multiple lesions. The tumor tends to be very locally aggressive, with infiltration of adjacent skeletal muscles, tendons, or periosteum, and erosion of bone. Approximately 12% of pediatric patients with desmoid fibromatosis have other congenital abnormalities.1 Adult-type intra-abdominal desmoid tumors are associated with familial adenomatous polyposis (FAP) and Gardner syndrome.1,17 Although most tumors in children are sporadic, there are reported cases of associated Gardner syndrome in childhood, one of which presented in infancy.18–20 The finding of minor radiologic bone abnormalities in 80% of patients with desmoids and 48% of their relatives suggests an autosomal dominant mode of inheritance.1 Antecedent trauma, including surgery or irradiation, is reported in 12–63% of patients with all forms of desmoid tumor.1 It is postulated that desmoid tumors are associated with a familial defect in the regulation of connective tissue and may be precipitated by multiple factors.1,17,21 Desmoid fibromas in adults and children can be associated with mutations in the adenomatosis polyposis coli (APC) or β-catenin CTNNB1 genes.22 The tumor is composed of bundles of slender, uniform spindle cells surrounded by variable amounts of collagen. Cleft or slit-like blood vessels are variable in number and more abundant at the periphery. The fibrous proliferation may be indistinguishable from scar tissue, except that it infiltrates skeletal muscle and tendons. Cellularity is variable. Some childhood lesions have an increased number of mitoses and greater cellularity.1 Immunohistochemical and ultrastructural studies show that the lesion is composed of fibroblasts and myofibroblasts. Immunohistochemistry and genotyping may help to distinguish sporadic from FAP cases.22 Myofibromatosis and other juvenile fibromatoses should be considered in the differential diagnosis. Keloid scars are more superficial than desmoid tumors. Cellular variants must be distinguished histopathologically from fibrosarcoma. Local excision with wide margins, if possible, is the mainstay of treatment.16–19,23 The recurrence rate varies from 30–80%.1 Higher recurrence rates are associated with a young age at diagnosis, large lesions, intralesional or marginal excision, and intra-abdominal location. Microscopic features of high vascularity, myxoid foci, and abundant immature myofibroblasts are associated with a higher recurrence rate.1 Treatment with combination chemotherapy and radiotherapy, or with tamoxifen and nonsteroidal anti-inflammatory agents, has been advocated for nonresectable, symptomatic or progressive disease.19,24 Mortality from locally aggressive desmoids is less than 10%.1 Fibromatosis colli, also known as sternocleidomastoid pseudotumor of infancy or congenital muscular torticollis, is a congenital fibromatosis of the sternocleidomastoid muscle. It occurs in up to 0.4% of live newborns.1 Males are affected more than females. It does not involve the skin. A hard, nontender, lobulated subcutaneous mass is palpable in the lower third of the sternocleidomastoid muscle. The trapezoid muscle is sometimes involved. Following an initial rapid period of growth, the tumor stabilizes in size. Torticollis and facial asymmetry are variable and may be transient. There is a right-sided predominance, and 2–3% of cases are bilateral.1 The pathogenesis is unknown. Birth trauma has been implicated in 50% of cases, with a history of complicated delivery; whether this is a cause or an effect of the tumor is not clear. Familial cases are rare.1 Histopathologically, bands of fibroblasts with abundant collagen are intermingled with residual angulated skeletal muscle fibers. The diagnosis may be established by fine-needle aspiration, which shows benign spindle cells and degenerating skeletal muscle fibers. Ultrasound or magnetic resonance imaging may also be useful.25 A combination of the typical location of the lesion in the neck and the characteristic histopathology is diagnostic. The clinical differential diagnosis of a neck swelling includes lymphatic malformation, hemangioma, and malignant neoplasms. The histopathologic features may resemble those of a desmoid tumor.1 The majority of lesions regress within the first year of life. Most resolve completely, but minor residual asymmetry or tightening of the sternocleidomastoid muscle is seen in 25% of cases.1 Only 9% have persistence of the tumor and torticollis. Physiotherapy is the treatment of choice.26 Surgery is rarely necessary unless the diagnosis is in doubt or the mass fails to resolve. Infantile digital fibromatosis is a recurring myofibroblastic proliferation of the fingers and toes. Synonyms for this tumor include digital fibrous tumor of Reye, digital fibrous swelling, recurring digital fibrous tumor of childhood, and inclusion body fibromatosis.1 Almost all lesions are diagnosed in infancy, and one-third are present at birth. Both sexes are affected equally. The typical lesion is an asymptomatic, firm, smooth, pink nodule located on the lateral or dorsal aspect of the digit, measuring <3 cm in diameter (Fig. 26.6). Lesions are more common on the fingers than on the toes. The thumbs and great toes are spared. There is often deformity of the affected digit. There may be single or multiple nodules (Fig. 26.7). Rarely, more than one digit is involved or extradigital lesions are seen.27 Periosteal attachment is not unusual, but underlying bone erosion is rare. The pathogenesis is not known. Defective organization of actin filaments in myofibroblasts has been hypothesized.1 Whorls and interdigitating sheets of uniform fibroblasts in a densely collagenous stroma are seen in the dermis or subcutis.1 A unique feature is the presence of distinctive, eosinophilic, perinuclear cytoplasmic inclusions surrounded by a clear halo that stain red with a trichrome stain. Electron microscopy shows abundant cytoplasmic filaments that form whorled bodies; these are the ultrastructural correlate of the cytoplasmic inclusions. Immunostaining is positive for desmin, actin, vimentin, and keratin. The digital location and the characteristic histopathology distinguish this lesion from other fibromatoses and pediatric soft tissue tumors. The local recurrence rate is 60–90% following surgical excision. Many tumors regress spontaneously within a few years.27 Conservative management without surgery is appropriate, unless there is functional impairment.27 Mohs micrographic surgery to debulk the tumor has been performed.28 Successful treatment with intralesional fluorouracil injections has been reported in a 7-year-old child.29 Fibrous hamartoma of infancy is a benign fibrous tumor that develops during the first 2 years of life.30 Up to 20% are present at birth.1 Occasional cases have been described in children between 2 and 10 years. Males are affected more frequently than females. Fibrous hamartoma presents as a subcutaneous lesion located around the axillae, shoulders, and upper chest wall.31 It may involve other sites, such as the inguinal region, extremities, and head and neck. It is usually a solitary nodule, measuring 2–5 cm in diameter, that feels lumpy to palpation. There may be overlying hypertrichosis. Occasionally, these lesions are multifocal. There are no symptoms. There are no systemic associations. Fibrous hamartoma of infancy is believed to represent a hamartomatous process rather than a true neoplasm. It is not familial. The hamartoma is located in the subcutaneous and musculoaponeurotic tissues.30,31 Histopathologic examination reveals three characteristic elements: a fibrous component consisting of well-defined fascicles of fibroblasts or disorderly fibroblasts in a collagenous stroma; mature adipose tissue; and myxoid mesenchymal tissue in a basophilic matrix. Electron microscopy reveals the presence of both fibroblasts and myofibroblasts, primitive mesenchymal cells, small blood vessels, and mature adipocytes.1 The clinical differential diagnosis of fibrous hamartoma of infancy includes a macrocystic lymphatic malformation, hemangioma, and other soft tissue tumors. Identification of the three histopathologic components of this lesion distinguishes it from other fibroblastic proliferations.31 A similar hamartoma with an admixture of fat and an absence of fibromyxoid tissue is described as lipofibromatosis.32 There is no tendency to spontaneous regression. The treatment of choice is surgical excision. The recurrence rate is low, even with incomplete excision.31 Lipofibromatosis is a rare, slowly growing tumor of fibroblasts and mature adipose tissue.1 It affects infants and young children and is congenital in up to 25% of cases. The tumor typically arises on the distal extremities and is less often encountered on the head and neck.1 Lesions range in size from 2–7 cm in diameter. Histopathologically, there is abundant adipose tissue traversed by fascicles of fibroblasts with variable collagen and focal myxoid change. Spindle cells stain positive for smooth muscle actin. The differential diagnosis of lipofibromatosis includes fibrolipoma, lipofibromatous hamartoma, fibrous hamartoma of infancy, lipoblastoma, and desmoid fibromatosis. Treatment is by surgical resection but the rate of recurrence or persistent growth is high. This is a rare familial disorder that manifests at the time of eruption of the deciduous or permanent teeth. There is slowly progressive gingival enlargement that may cover the crowns of the teeth and result in difficulty in eating or speaking. It is associated with generalized hypertrichosis. Rarely there is associated mental retardation and epilepsy. The Zimmerman–Laband syndrome is characterized by gingival fibromatosis, hypertrichosis, intellectual disability, and absence and/or hypoplasia of the nails or terminal phalanges of the hands and feet as well as other anomalies.33 Mucosal biopsy shows coarse, interlacing collagen bundles with sparse fibroblasts and myofibroblasts. There may be calcification, ossification, abundant amorphous extracellular material, and cellular fibroblastic proliferation. The differential diagnosis includes phenytoin usage, chronic gingivitis, cherubism, juvenile hyaline fibromatosis, and other rare syndromes. Treatment options include repeated surgical debulking of the gums or dental extraction. The superficial fibromatoses of adulthood are the most common type of fibromatosis in the general population but are rare in infants and children. Fibromatosis may involve the palm (Dupuytren contracture), the plantar surface of the foot (Ledderhose disease), or the penis (Peyronie disease). Dupuytren-type fibromatosis of the palms and soles may be seen in childhood, and is occasionally congenital (Fig. 26.8 Leiomyoma is a benign tumor of smooth muscle. Cutaneous leiomyoma may arise from the arrector pili muscle in hair follicles, the dartos muscle of the scrotum and labia majora, the erectile muscle of the nipple, and the muscular wall of veins (angioleiomyoma). Leiomyomas are uncommon in children and are extremely rare in the newborn period.37 Cutaneous leiomyomas appear as discrete papules or nodules with a pink or brown discoloration of the overlying skin. They are usually solitary but may be multiple. Rarely, a leiomyoma may present as a pedunculated mass at birth or as a papular plaque in early infancy.37–39 Leiomyomas are often painful, particularly on exposure to cold. Leiomyomas of the tongue are also reported.38 Most cutaneous leiomyomas are not associated with visceral disease. The multiple leiomyomas of the esophagus and tracheobronchial tree in Alport syndrome may be associated with female genital leiomyomas in older children and adults.40 Leiomyomas that occur in immunocompromised children only rarely involve the skin or soft tissues.41 Multiple cutaneous leiomyomas presenting in adolescence or adult life may be inherited as an autosomal dominant trait, but the etiology of other forms is unknown. The diagnosis is made by skin biopsy, which demonstrates whorls and bundles of well-differentiated spindle cells with cigar-shaped nuclei in the dermis. There is a variable collagenous component. The smooth muscle stains red with the Masson trichrome stain. Immunochemistry is positive for muscle-specific actin and desmin reactivity. Leiomyoma must be distinguished from the fibroblastic and myofibroblastic proliferations of infancy and childhood, as well as from other spindle cell tumors such as neurofibroma and leiomyosarcoma. Immunohistochemistry may be helpful, as myofibroblastic tumors express smooth muscle actin more than muscle-specific actin or desmin.37 The circumscribed spindle cell appearance of leiomyoma differs from the smooth muscle bundles of congenital smooth muscle hamartoma. Excision is curative for solitary lesions. Cutaneous neurofibromas in infants and young children are most frequently associated with neurofibromatosis type 1 (NF-1) (see Chapter 29). These benign tumors consist of Schwann cells, nerve fibers, and fibroblasts, and may be cutaneous, subcutaneous, or plexiform. Cutaneous and subcutaneous neurofibromas are rarely seen at birth but may sometimes appear within the first year of life. Plexiform neurofibromas are often present at birth and are considered pathognomonic of neurofibromatosis. There may be a large area of hyperpigmentation overlying the plexiform neurofibroma that predates the characteristic ‘bag of worms’ consistency of the tumor. These lesions enlarge with time and can cause considerable cosmetic disfigurement, particularly on the face and around the eye. A plexiform neurofibroma in the neck may compromise airway function, and large lesions over the back are often associated with underlying spinal involvement. Schwannomas are found in both neurofibromatosis type 2 (NF-2) and in schwannomatosis. Schwannomatosis, or neurilemmomatosis, is characterized by multiple cutaneous schwannomas and can present at birth or develop during childhood.42 These lesions are a marker for development of central nervous system tumors in later childhood and adult life. Schwannomatosis is sometimes associated with SMARCB1 mutations and is distinguished from NF-2 primarily by the absence of vestibular tumors.42 Pacinian neurofibromas, or nerve-sheath myxomas, are uncommon tumors with components that resemble Vater–Pacini corpuscles.43 Multiple hairy pacinian neurofibromas have been reported in children without NF-1 and may be congenital.43 Underlying skeletal anomalies may be associated with pacinian neurofibromas in a sacrococcygeal location. The non-Langerhans’ cell histiocytoses encompass a diverse group of disorders in which there is proliferation of mononuclear phagocytes other than Langerhans’ cells. Two variants, juvenile xanthogranuloma and benign cephalic histiocytosis, occur primarily in infants and young children. Other benign histiocytoses, such as papular xanthoma, xanthoma disseminatum, and generalized eruptive xanthoma, may rarely present in childhood, but are extremely unusual in infancy.44,45 Juvenile xanthogranuloma is a benign, self-healing, non-Langerhans’ cell histiocytosis characterized by solitary or multiple yellow-red papules and nodules in the skin and occasionally in other organs.46 Although adults may be affected, it is predominantly a disorder of infancy and early childhood. There is an increased frequency of juvenile xanthogranuloma in children with NF-1, juvenile myeloid leukemia, and urticaria pigmentosa.47,48 Juvenile xanthogranulomas have also developed in children with Langerhans’ cell histiocytosis either concurrently or at a later date.49 There are single case reports of coexistence with Wiskott–Aldrich syndrome and acute lymphoblastic leukemia.50,51 The typical juvenile xanthogranuloma is an asymptomatic, firm, well-demarcated papule or nodule that measures from 1 mm to 2 cm in diameter. Early lesions are pink or red in color, later changing to a distinctive yellow or orange-brown (Fig. 26.9). There may be overlying telangiectasia with a purpuric appearance, and occasionally surface ulceration and bleeding with associated pruritus and discomfort. Solitary lesions with a hyperkeratotic surface, pedunculated or plaque-like morphology are also reported.46 As many as 17% of juvenile xanthogranulomas are present at birth, and 70% develop within the first year of life.46 The majority are solitary lesions. Multiple lesions may be few or number in the hundreds (Fig. 26.10A). They can be located at virtually any body site, but are most common on the head, neck, and upper trunk. Juvenile xanthogranulomas may be classified as micronodular, measuring 2–5 mm, or macronodular, measuring 0.5–2 cm in diameter. An unusual variant is the giant juvenile xanthogranuloma, which measures from 2–10 cm in diameter (Fig. 26.10B).52 These lesions are congenital or appear in early infancy and may have a greater propensity to ulcerate. Rarely, numerous micronodular lesions may present as a generalized lichenoid eruption.53 Extracutaneous juvenile xanthogranuloma is rare, and less than 50% of these patients have associated cutaneous lesions.54 The most frequent extracutaneous sites are the eye and orbit, central nervous system, liver/spleen, lung, oropharynx, and muscle. In contrast to cutaneous lesions, a systemic juvenile xanthogranuloma may produce symptoms related to a mass effect or infiltration of the involved organ. The incidence of ocular disease in patients with cutaneous lesions is 0.3–0.4%.55 Eye lesions manifest as an asymptomatic mass on the iris, unilateral glaucoma, spontaneous hyphema, or color change of the iris. Risk factors for eye involvement include multiple lesions, age <2 years, and recently diagnosed disease.55 Juvenile xanthogranulomas are seen with increased frequency in patients with NF-1. A triple association between juvenile chronic myeloid leukemia, juvenile xanthogranulomas, and NF-1 is recognized (Fig. 26.11 The typical histopathologic appearance consists of a dense dermal infiltrate of foamy histiocytes with Touton giant cells. There is an admixture of other cell types, including lymphocytes, eosinophils, neutrophils, and foreign body giant cells, as well as infrequent mitoses. In early lesions, there may be few or absent foam cells or Touton giant cells, with a variable number of spindle cells and numerous mitotic figures.59 Immunohistochemistry shows negative staining for S100 and CD1a, and positive staining for factor X111a, CD68, CD163, fascin and CD14.56 There are no Birbeck granules visible on ultrastructural examination. Distinguishing a small juvenile xanthogranuloma from clinically similar lesions such as xanthoma, mastocytoma, Spitz nevus, and other benign skin tumors may require a skin biopsy. Giant lesions may be mistaken for a hemangioma or malignant tumor. Early lesions that lack the characteristic lipid-laden histiocytes and Touton giant cells may resemble Langerhans’ cell histiocytosis on histopathologic examination.56,59 The absence of Birbeck granules and negative staining for S100 is characteristic of juvenile xanthogranuloma. Most cutaneous lesions resolve spontaneously over months or years and do not require treatment. Ulcerating, symptomatic, or large unsightly lesions may require surgical excision. A residual area of hyperpigmentation or anetoderma-like atrophy may persist. Ocular and systemic lesions can be more problematic and involvement of the liver and bone marrow can be life-threatening.60 Treatment options include observation, corticosteroids, surgical excision, radiation therapy, and chemotherapy.46,60 Benign cephalic histiocytosis is a non-Langerhans’ cell histiocytosis characterized clinically by multiple brownish-yellow macules and papules on the face and adjacent areas. Some authors believe that benign cephalic histiocytosis is a variant of micronodular juvenile xanthogranuloma.61,62 Lesions first appear between the ages of 2 months and 2 years. The face is the site of predilection, but the scalp, neck, and ears can also be involved. Lesions may be scattered over the shoulders and upper arms. Typical lesions are slightly raised, asymptomatic papules measuring 2–3 mm in diameter that vary from erythematous to light-brown or yellowish in color (Fig. 26.12 Typically, extracutaneous disease is absent, but there has been one report of associated diabetes insipidus.63 Histopathologically, a monomorphous histiocytic infiltrate is located in the upper and mid-dermis. There may also be a few lymphocytes and eosinophils. Foamy macrophages and Touton giant cells are typically absent. Staining for S100 protein is usually negative.64 Electron microscopy reveals coated vesicles and comma- or worm-shaped bodies. Birbeck granules are absent. The differential diagnosis includes juvenile xanthogranuloma, Langerhans’ cell histiocytosis, and cutaneous mastocytosis. The lesions of mastocytosis have a similar color but urticate when rubbed (Darier sign) and have a distinctive histology. Benign cephalic histiocytosis can be distinguished from Langerhans’ cell histiocytosis by immunohistochemical stains and the absence of Birbeck granules on electron microscopy. There is no effective treatment. The skin lesions regress spontaneously over months to years. There may be residual hyperpigmentation and anetoderma-like atrophy. Calcium deposition in the skin, or calcinosis cutis, is found in a diverse group of disorders. It is termed dystrophic calcification when calcium is deposited in abnormal or injured tissue in patients with no abnormality of calcium or phosphate metabolism. Metastatic calcification develops in normal tissues as a result of abnormal calcium and phosphorus metabolism. Idiopathic calcification occurs in the absence of any discernible tissue injury or metabolic abnormality. Iatrogenic calcification may develop as a complication of calcium infusions or the application of calcium-containing paste to abraded skin. Cutaneous ossification, in which normal bone is formed in the dermis and subcutaneous soft tissues, is termed osteoma cutis. Dystrophic calcification arises at sites of skin trauma or in association with inflammatory lesions, connective tissue disorders, skin tumors, and cysts. Calcinosis cutis on the heels is a not uncommon sequela of drawing blood by heel sticks during the neonatal period.65,66 It presents some months later as one or more white papules or nodules, and usually resolves spontaneously by 18–30 months of age. Calcification may also occur in association with subcutaneous fat necrosis of the newborn.67,68 Calcium deposition has been observed histopathologically both in the septa and within the fat lobules.67,69 Widespread subcutaneous calcification may develop in cases of subcutaneous fat necrosis complicating hypothermic cardiac surgery.68–70 Although hypercalcemia is a known complication of subcutaneous fat necrosis, the majority of reported cases of soft-tissue calcification have occurred in normocalcemic patients. Conversely, most infants with subcutaneous fat necrosis and hypercalcemia do not show evidence of calcium deposition in biopsies taken from affected sites.68 Dystrophic calcification has been reported in the skin lesions of a newborn infant with intrauterine-acquired herpes simplex infection.71 The calcification was present at birth and appeared to have developed in utero. A lethal disorder characterized by extensive congenital skin necrosis and follicular calcification has been described in three newborn females.72 Dystrophic calcification may also occur as a complication of intralesional corticosteroid injection of infantile periocular hemangiomas.73 Metastatic calcification occurs when calcium salts are precipitated in normal tissues as a result of high serum calcium or phosphate levels. The calcium deposits usually consist of hydroxyapatite crystals. This is associated primarily with chronic renal insufficiency, in which ulceration of the skin may be caused by calcification of blood vessels, leading to ischemic skin necrosis, or by painful disseminated calcification of the dermis and subcutaneous tissues (calciphylaxis).74 Chronic renal failure is also associated with benign nodular calcification. Cutaneous calcium deposits may develop as a result of hypervitaminosis D, milk-alkali syndrome, and other causes of hypercalcemia and hyperphosphatemia. Metastatic calcinosis in the skin is rarely seen in infancy and childhood.75 In contrast, the cutaneous bone formation, or osteoma cutis, associated with Albright hereditary osteodystrophy frequently appears first in infancy or childhood and may present in the neonatal period. This metabolic disorder is discussed below. Idiopathic calcification can be congenital or acquired. Congenital calcified nodules occur most frequently on the ear, but may be seen elsewhere on the face and limbs. These lesions are variously described as congenital calcified nodule of the ear, subepidermal calcified nodule, or solitary congenital nodular calcification of Winer.76–78 Other types of idiopathic calcinosis cutis, such as idiopathic calcification of the scrotum or vulva, and the milia-like lesions associated with Down syndrome, present later in childhood or adolescence and are not seen in the newborn. There are rare reports of juxta-articular tumoral calcinosis in infancy.79–82 A solitary calcified nodule on the pinna or earlobe is the most common presentation of idiopathic calcinosis in the newborn (Fig. 26.13). These nodules may occur elsewhere on the face or limbs, and occasionally there is more than one. Auricular lesions developing after birth have also been described. There is a male preponderance. The nodule is firm and measures 3–10 mm in diameter. The surface may be warty in appearance, or smooth and dome-shaped. The color is chalky white or yellow. Surface ulceration and discharge of calcified material may occur spontaneously or as a result of trauma. There are usually no associated symptoms. Serum calcium and phosphate levels are normal. There are no systemic abnormalities. The pathogenesis of these lesions is not clear. Most authors believe they represent dystrophic calcification following dermal damage from some unknown source. Proposed hypotheses include derivation from milia, syringomas, other sweat gland hamartomas, nevi, trauma, and ischemic injury.77,78 The diagnosis is often made on the clinical appearance. Histopathologically, amorphous and/or globular masses of calcified material are seen in the papillary dermis and may extend to the reticular dermis. Foreign body giant cells may be observed in association with the calcified masses. The overlying epidermis shows a warty architecture with variable amounts of pseudoepitheliomatous hyperplasia. Ulceration and transepidermal elimination of calcium may occur. Clinically, calcified nodules may be misdiagnosed as viral warts, molluscum contagiosum, pilomatricomas, syringomas, and congenital inclusion cysts. If treatment is necessary, the nodule can be removed by curettage or excision. Calcified nodules sometimes recur following curettage or shave excision. Intralesional injection of triamcinolone at the time of shave excision has been suggested for recurrent lesions.76 Tumoral calcinosis is characterized by painless, calcified soft tissue nodules located close to large joints in otherwise healthy children and adults.79–82
Lumps, Bumps, and Hamartomas
Lumps and bumps
Fibromatoses
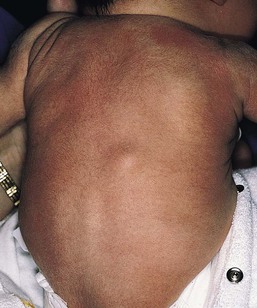
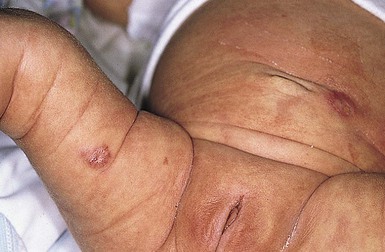
Infantile myofibromatosis
Cutaneous findings.
Extracutaneous findings.
Etiology and pathogenesis.
Diagnosis.
Differential diagnosis.
Treatment and prognosis.
Infantile desmoid-type or aggressive fibromatosis
Clinical findings.
Etiology and pathogenesis.
Diagnosis.
Differential diagnosis.
Treatment and prognosis.
Fibromatosis colli
Clinical findings.
Etiology and pathogenesis.
Diagnosis.
Differential diagnosis.
Treatment and prognosis.
Infantile digital fibromatosis
Cutaneous findings.
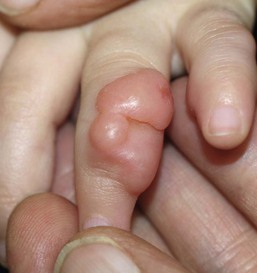
Extracutaneous findings.
Etiology and pathogenesis.
Diagnosis.
Differential diagnosis.
Treatment and prognosis.
Fibrous hamartoma of infancy
Cutaneous findings.
Extracutaneous findings.
Etiology and pathogenesis.
Diagnosis.
Differential diagnosis.
Treatment and prognosis.
Lipofibromatosis
Gingival fibromatosis
Cutaneous findings.
Extracutaneous findings.
Diagnosis.
Differential diagnosis.
Treatment and prognosis.
Adult-type fibromatoses
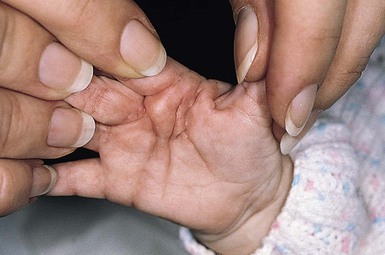
 ).1 Surgical excision is only necessary for diagnosis or for release of contractures. Knuckle pads are seen in older children and adolescents but not in infants.
).1 Surgical excision is only necessary for diagnosis or for release of contractures. Knuckle pads are seen in older children and adolescents but not in infants.
Leiomyoma
Cutaneous findings.
Extracutaneous findings.
Etiology and pathogenesis.
Diagnosis.
Differential diagnosis.
Treatment and prognosis.
Neurofibromas and other neural tumors
Non-langerhans’ cell histiocytoses
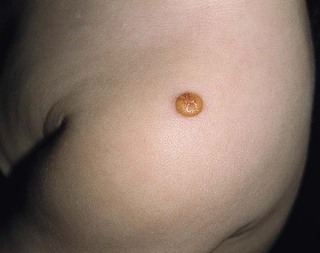
Juvenile xanthogranuloma
Cutaneous findings.
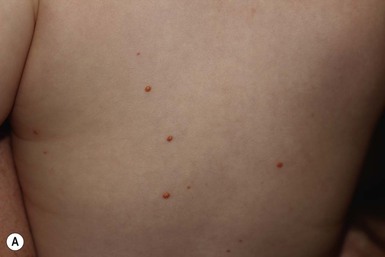
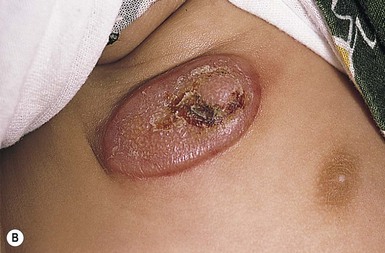
Extracutaneous findings.
 ).47
).47
Diagnosis.
Differential diagnosis.
Treatment and prognosis.
Benign cephalic histiocytosis
Cutaneous findings.
 ). The mucous membranes are not involved.
). The mucous membranes are not involved.
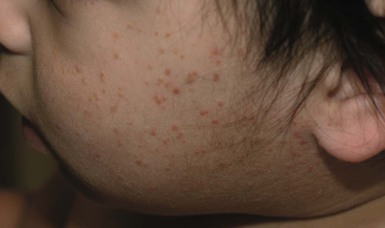
Extracutaneous findings.
Diagnosis.
Differential diagnosis.
Treatment and prognosis.
Calcifying disorders of the skin
Dystrophic calcification
Metastatic calcification
Idiopathic calcification
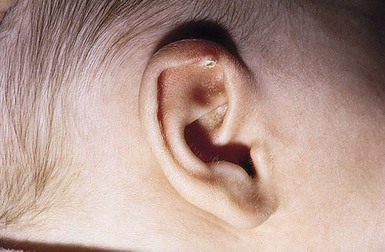
Calcified ear nodule
Cutaneous findings.
Extracutaneous findings.
Etiology and pathogenesis.
Diagnosis.
Differential diagnosis.
Treatment and prognosis.
Tumoral calcinosis
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Lumps, Bumps, and Hamartomas
26