Fig. 3.1
Overview of epidermal keratinocytes in innate immunity. The keratinocyte is the first cell to come in contact with and sense pathogens. Keratinocytes detect gram-positive bacteria and virus-associated double-stranded RNA (ds-RNA) through Toll-like receptor (TLR) 2 and TLR3, respectively. Keratinocytes express the intracellular sensors NOD1/2 and recognize g-D-glutamyl-meso-dianopimelic acid (iE-DAP) and muramyl dipeptide (MDP). β-glucan can be detected by dectin-1. After pathogen recognition, keratinocytes produce various cytokines, chemokines, and antimicrobial proteins (AMPs). Group 1 house dust mite (HDM) allergens activate the NLRP3 inflammasome of keratinocytes
Although the epidermal keratinocytes are in constant contact with the pathogens on the skin surface, they do not induce inflammation. This lack of inflammation can be explained partially by the presence of Staphylococcus epidermidis on the epidermal surface because lipoteichoic acid derived from S. epidermidis reduces the TLR3-induced skin inflammation through TLR2 on epidermal keratinocytes [10].
3.2 AMPs
Various epithelial cells produce AMPs to protect against pathogens [11]. AMPs are members of diverse protein families including defensins, cathelicidins, C-type lectins, ribonucleases, and S100 proteins. Cathelicidin is cleaved to generate the active form, LL-37. Keratinocytes, the main source of AMPs in the epidermis, express human β-defensins (hBD) 1–4 and cathelicidin. Additionally, hBDs and cathelicidin are produced by the large intestine, respiratory tract, and urinary tract. The cationic hBDs and LL-37 interact with the negatively charged phospholipids in the pathogens, which is followed by insertion in the membrane of the pathogen and disruption of the cell wall (Fig. 3.2). Whereas expression of cathelicidin and hBDs remain low in the steady-state condition, expression increases markedly upon infection, inflammation, or presence of wounds [3, 11]. These peptides have antimicrobial activity against diverse pathogens including gram-positive and -negative bacteria, fungi, and viruses. However, methicillin-resistant S. aureus (MRSA ) is resistant to hBD3 and LL-37 [12], which may explain the high susceptibility of MRSA in skin wounds. Although these peptides have been identified as antibacterial molecules, they have various biological activities. For example, LL-37 causes keratinocyte migration and angiogenesis (Fig. 3.1), which may accelerate wound closure [11, 13].
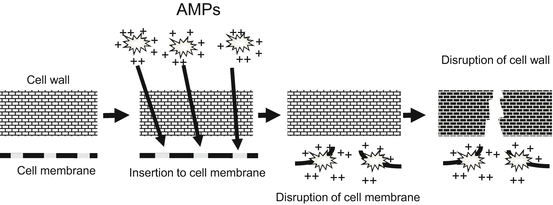
Fig. 3.2
AMP mechanism of action. Cationic human β-defensins (hBDs) and LL-37 (the active form of cathelicidin) interact with negatively charged phospholipids on the pathogens; this interaction leads to insertion into the membrane of the pathogen and disruption of the cell wall
3.3 Allergy Triggered by Keratinocytes in Innate Immunity
Epidermal keratinocytes function in innate immunity of the skin. On the other hand, the innate immunity function of epidermal keratinocytes is a key for triggering or exacerbating allergic diseases such as atopic dermatitis (AD).
3.3.1 Allergy Triggered by S. aureus
AMPs control the skin surface pathogens including S. aureus . However, AMP expression is decreased in AD [14], which causes colonization of S. aureus on the skin. Although S. aureus is not an allergen, this colonization on the skin is an exacerbating factor for AD (Fig. 3.3). Thymic stromal lymphopoietin (TSLP) is an epithelium-derived cytokine essential for generating skin allergic inflammation and the Th2 type response. TSLP expression is increased in the epidermal keratinocytes affected by AD [15]. S. aureus acts directly on the keratinocytes through TLR2/6 to release TSLP [16]. Increased TSLP expression promotes allergen sensitization, which ultimately leads to AD.
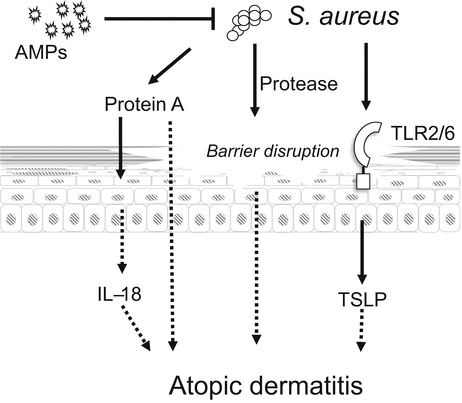
Fig. 3.3
Staphylococcus aureus is an exacerbating factor for atopic dermatitis (AD). AMP expression is decreased in AD, which causes colonization of S. aureus on the skin. S. aureus acts directly on keratinocytes through TLR2/6 to release thymic stromal lymphopoietin (TSLP). Protein A derived from S. aureus induces IL-18 release from keratinocytes. Furthermore, S. aureus protease disrupts the epidermal barrier function
Mice treated with repeated applications of S. aureus protein A develop AD-like skin inflammation [17], and protein A of S. aureus acts on keratinocytes to release IL-18 [18]. Furthermore, the extracellular protease of S. aureus causes epidermal barrier dysfunction [19], which allows allergens or pathogens to penetrate into the epidermis and stimulate keratinocytes.
Therefore, direct interaction of S. aureus with keratinocytes is a key event in the pathogenesis of AD.
3.3.2 AD and House Dust Mite (HDM) Allergen
The pathogenesis of AD involves the interactions of multiple factors including susceptibility genes, environmental factors, skin barrier defects, and immunological factors [20]. Among these, skin barrier dysfunction is one of the major manifestations of AD and a key contributor to its pathogenesis. Barrier disturbances resulting from genetic defects or abnormal expression of epidermal proteins caused by Th2-type cytokines may increase the risk of exposure to allergens and contribute to the development of AD. However, the mechanisms underlying the relationship between immunological and physical defects remain unclear. Among the environmental factors, HDM allergens are important for the development of AD, asthma, and rhinitis [21]. Barrier defects may allow epidermal penetration of HDM allergens and increase the likelihood of allergen exposure to epidermal keratinocytes. This direct contact between HDM allergens and keratinocytes is important for triggering inflammation because the innate immunity of keratinocytes is activated by the HDM allergens [22].
3.3.3 NLRP3 Inflammasome
NLR members form an inflammasome, a multiprotein complex that activates caspase-1, which ultimately leads to the processing and release of the proinflammatory cytokines IL-1β and IL-18. Growing evidence supports the idea that inflammasomes play important roles in skin inflammation, especially the NLRP3 inflammasome (also known as NALP3 or cryopyrin), which comprises oligomerized NLRP3, apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC), and caspase-1 (Fig. 3.4). Stimulated caspase-1, expressed initially as an inactive precursor, is activated to cleave IL-1β and IL-18 proforms. The inflammasome can be activated not only by pathogen-associated molecules but also by various other stimuli including the “danger-associated molecular pattern” molecules (such as ATP and urate crystals), environmental stimuli (such as silica crystals and aluminum salt) [23], and neurodegenerative stimuli (such as amyloid-β fibrils) [24].
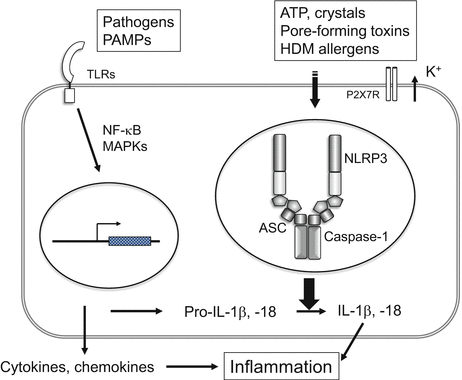
Fig. 3.4
NLRP3 inflammasome. Activation of the NLRP3 inflammasome requires two signals: (1) Pathogens or pathogen-associated molecular patterns (PAMPs) increase the expression of the IL-1β and IL-18 proforms through TLRs; and (2) ATP, crystals, bacterial pore-forming toxins, or HDM allergens activate the NLRP3 inflammasome. The NLRP3 inflammasome comprises oligomerized NLRP3, apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC) and caspase-1. Caspase-1 is expressed initially as an inactive precursor; following stimulation, activated caspase-1 cleaves IL-1β and IL-18 proforms
3.3.4 Activation of the Keratinocyte Inflammasome by Group 1 HDM Allergens
Dermatophagoides pteronyssinus (Der p) and D. farinae (Der f) are the most common types of HDM in temperate climates. The group 1 allergens, which exhibit cysteine protease activity, activate caspase-1 and induce the caspase-1-dependent release of IL-1β and IL-18 from keratinocytes [22]. The group 1 allergens assemble inflammasomes by recruiting ASC, caspase-1, and NLRP3 to the perinuclear region (Fig. 3.5) [22]. The group 1 allergens are novel effective activators of the inflammasome in epidermal keratinocytes. Recent studies using mouse models have indicated the importance of IL-18 for the development of AD [17, 25]. IL-18 regulates allergic inflammation by inducing Th2 cytokine production and eosinophilia; therefore, HDM-induced IL-18 release from keratinocytes might be important in the pathogenesis of AD.
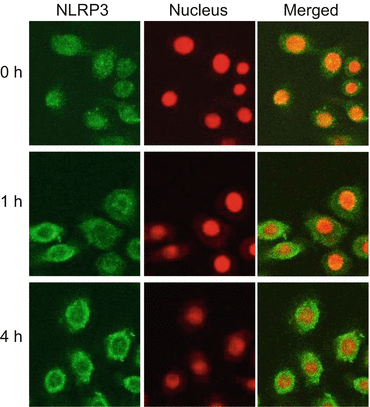
Fig. 3.5
Formation of NLRP3 inflammasomes in keratinocytes. Cultured keratinocytes were stimulated with house dust mite (HDM) allergens. Subcellular distribution of NLRP3 was assessed with immunofluorescence. Staining for NLRP3 revealed diffuse distribution throughout the resting cells (0 h). After 1 h of stimulation, NLRP3 translocated rapidly to the cytosol and accumulated in the perinuclear region. ASC and caspase-1 were also translocated to the perinuclear region [22], indicating that formation of the NLRP3 inflammasome was induced by the HDM allergen (Adopted from Ref. [22])
3.3.5 Activation of Keratinocyte by Group 2, 3, and 9 HDM Allergens
As do class 1 HDM allergens, class 2, 3, and 9 HDM allergens also activate keratinocytes (Fig. 3.6). The class 2 allergens are major antigens for IgE, and lack protease activity [26]. Unlike class 1 HDM allergens, class 2 HDM allergens do not induce the release of IL-1β from keratinocytes, but stimulate IL-8 release via a nonproteolytic mechanism [22], which presumably involves the activation of NF-κB and MAPK signaling pathways [27, 28]. Class 2 HDM allergens activate airway smooth muscle cells in a TLR2/MyDD88-dependent manner [28]; therefore, class 2 HDM allergen-induced keratinocyte activation may involve the TLR2 pathway. Class 3 and 9 HDM allergens, which have serine protease activity, induce the release of IL-8 and GM-CSF from keratinocytes [29] and airway epithelial cells [30] by activating protease-activated receptor 2 (PAR-2) signaling. Thus, keratinocytes are able to detect HDM allergens directly and initiate local inflammatory responses, perhaps through the cooperation of the NLRP3 inflammasome, PAR2, and even the TLR pathways.
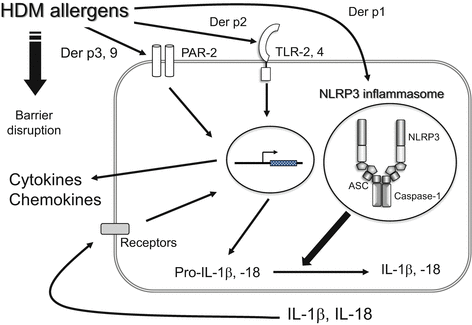
Fig. 3.6
Biological activity of HDM allergens. Each group of HDM allergens activates NLRP3 inflammasome, TLR-2,4 and PAR-2, respectively, resulting in the production of cytokines and chemokines. On the other hand, HDM protease disrupts the epithelial barrier
3.4 Allergen Activation of Epithelial Cells
The underlying mechanism by which particular molecules, such as HDM allergens, are recognized by hosts as “allergens” is largely unknown. Because epithelial cells are the first cells to come in contact with the allergens, the allergen-activation of the innate immunity of epithelial cells may be important for allergens to function as “allergens”.
The airway epithelial cells respond to HDM allergens as well as epidermal keratinocytes [30, 31]. In an allergic reaction in the airway, recognition of the HDM allergen by TLR4 on airway epithelial cells, rather than dendritic cells, is important [32]. Because Der p 2 has structural homology with the adaptor protein of TLR4 (MD-2), Der p 2 is involved in the LPS response by facilitating signaling through direct interactions with the TLR4 complex and reconstituting LPS-driven TLR4 signaling [33] (Fig. 3.6).
Ragweed pollen is one of the major allergens of seasonal rhinitis and conjunctivitis and is an exacerbating factor for AD. Similar to HDM allergens, ragweed pollen activates the keratinocyte NLRP3 inflammasome. Furthermore, in seasonal rhinitis, direct contact of ragweed pollen to nasal epithelial cells induces IL-33 release from the cells into the nasal fluid [34], which induces Th2 cytokine-mediated allergic inflammation.
All epidermal keratinocytes, airway epithelial cells, and nasal epithelial cells respond to allergens. Therefore, the direct contact and recognition of allergens by epithelial cells is important for allergens to function as “allergens” in the pathogenesis of allergic diseases such as AD, asthma, and seasonal rhinitis.
3.5 Protease Allergens and Epithelium
HDM allergens and ragweed pollen are protease allergens that increase the risk of sensitization to allergens. HDM allergens decrease the barrier function of the epidermis (Table 3.1) [35] and delay the recovery of the epidermal barrier [36]. Furthermore, group 1 HDM allergens disrupt intercellular tight junctions (TJs), which are the principal components of the epithelial permeability barrier [37]. Cleavage of TJs involves proteolysis of the TJ proteins, occludin, and ZO-1 [38]. Upon barrier disruption, HDM allergens gain increased access to the epithelial cells and can directly activate the epithelial cells. Similar barrier disruptions occur by other protease allergens such as pollen, aspergillus, and cockroach [36, 39]. Taken together, these protease allergens could function as “allergens” by disrupting the epithelial barrier to increase accessibility to the epithelial cells. This activation of epithelial cells may initiate a cascade of antigen recognition and allergic inflammation.
Table 3.1




Activity of allergen protease on epithelium
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
