, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
With special contribution from Andrea L. Haws, MD, The Children’s Hospital of San Antonio.
4.1 Lichen Planus
4.1.1 Clinical Features
Lichen planus most commonly occurs in individuals between 30 and 60 years old with children only accounting for 1–4 % of all cases [1]. Familial disease accounts for anywhere between 1 % and 50 % of cases, and may be seen more frequently in children [1]. Patients with a positive family history tend to present with disease at a younger age (median age at diagnosis of 26 years in familial disease versus 46 years in non-familial disease), and there is a greater likelihood of generalized involvement and oral mucosal findings as well as relapsing and prolonged disease course.
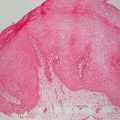
The physical exam findings of lichen planus are classically described as polygonal, purple (violaceous), flat-topped papules with overlying white reticulate scale (Wickham striae) (Fig. 4.1). Although nail involvement can be seen in up to 10 % of adult cases, it is an infrequent feature in children. Nail changes are characterized by thinning of the nail plate, longitudinal ridging, fragility, onycholysis, and pterygium formation [1]. Oral lesions are seen in 30–70 % of adult cases, and may be erosive with reticulate white patterning on the buccal and lingual mucosa . The incidence of oral disease in children is much less common with a prevalence between 4 % and 30 %. On the contrary, hypertrophic variants with hyperkeratotic plaques and adherent scale localized to the distal legs and ankles most often occur in children. Most active lesions resolve within 1 year, although post-inflammatory hyperpigmentation is noted in almost all cases. Hypertrophic lesions are more likely to resolve with residual dyschromia, atrophy, and scarring.
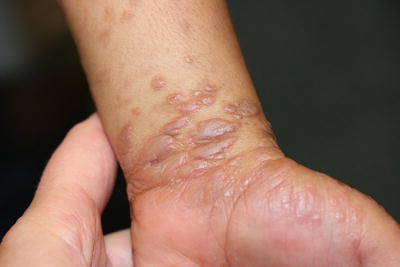
Fig. 4.1
Lichen planus is characterized by polygonal violaceous papules with overlying white reticulations often at the distal extremities
4.1.2 Histology
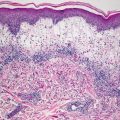
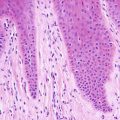
Lichen planus occurring in children has similar histologic features to the same disease in adults. The stratum corneum is hyperkeratotic without acanthosis. The epidermis is acanthotic with hypergranulosis (Fig. 4.2). The rete ridges demonstrate a saw-toothed alteration in appearance. The basal layer is disrupted and in some places obscured by a dense, band-like infiltrate of lymphocytes along the dermal epidermal junction (Fig. 4.3). Dying keratinocytes are present either focally or extensively, resulting in separation of the epidermis from the dermis. In some cases, this may present with clinically apparent bullae (bullous lichen planus) [2]. In older lesions, especially in patients with darker skin tones, post-inflammatory pigment incontinence is detected in the presence of papillary dermal melanophages (lichen planus pigmentosus in its most extensive form). The inflammatory infiltrate consists of lymphocytes and histiocytes. Eosinophils and plasma cells are not common. The infiltrate is restricted to the papillary dermis, and does not ordinarily extend into the reticular dermis [3–5].
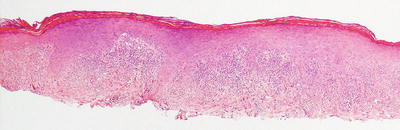
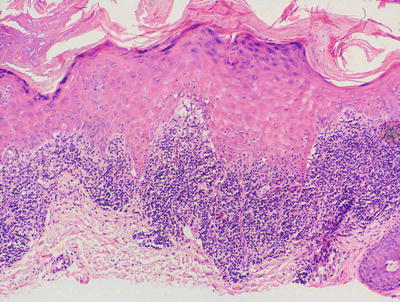
Fig. 4.2
Lichen planus has orthokeratotic hyperkeratosis, hypergranulosis, acanthosis and a bandlike lymphocytic infiltrate in the papillary dermis
Fig. 4.3
In some cases of lichen planus, the epidermis appear papillomatous and hypergranulotic. Scattered dying keratinocytes are seen along with a dense band-like infiltrate and focal exocytosis of lymphocytes into the epidermis
Actinic lichen planus consists of lesions with similar histology in a photo-distributed area. As this variant of the disease is more common in darker skinned individuals, dermal melanophages are frequently part of the histologic constellation of findings [6].
Oral lichen planus demonstrates similar histologic features with several differences [7]. These changes are more site-specific than anything about the disease itself. Parakeratosis is often present, since this is the normal finding in mucosal surfaces and not a feature of cutaneous lichen planus. The inflammatory infiltrate may contain plasma cells, again more a feature of the normal inflammatory response within the oral mucosa rather than lichen planus itself.
The histologic differential diagnosis is quite broad. Lupus erythematosus is characterized by epidermal atrophy and not acanthosis in most cases. The inflammatory infiltrate centers on cutaneous appendages and extends deep into the dermis, features not associated with lichen planus. Plasma cells may be present in the infiltrate, and dermal mucin is a helpful finding in lupus erythematosus. Lichen striatus may also present some diagnostic difficulty, but as with lupus erythematosus, the inflammatory infiltrate extends deep into the dermis and is commonly peri-eccrine. Moreover, the inflammatory response along the epidermal junction is often not quite as intense as is the case with lichen planus. Pityriasis lichenoides , both the acute and chronic variants, demonstrate overlying parakeratosis, which is not a feature of lichen planus. Additionally, in most cases of pityriasis lichenoides, the infiltrate is less band-like, extending deeper into the dermis in most cases, and hemorrhage is frequently encountered. Lichenoid drug eruptions may be histologically indistinguishable from lichen planus and require history to make the correct diagnosis in some situations. The presence of a deeper infiltrate, and eosinophils and plasma cells in the dermis would favor drug-induced lichenoid dermatitis over lichen planus.
Lichen planus pemphigoides is generally believed to be a heterogeneous group of disorders that share histologic features of lichen planus and bullous pemphigoid with linear deposition of IgG and C3 along the dermal-epidermal junction [4, 8]. Whether this entity is related to lichen planus remains controversial, but it remains in the histologic differential diagnosis on account of its similarities to lichen planus.
4.1.3 Pathogenesis
Lichen planus is a T cell-mediated autoimmune response. Both CD4+ and CD8+ T lymphocytes are present in the dermis, while CD8+ T lymphocytes infiltrate the epidermis in lichen planus lesions [9]. Most of the lymphocytes in the infiltrate are CD8+ and CD45RO+ cells and express the α-β T cell receptor (TCR) . These cells are believed to trigger apoptosis of basal keratinocytes in the epidermis that is often observed in a lichenoid inflammatory reaction [10]. Possible mediators of keratinocyte cell death include perforin and granzyme B [11].
Several cytokines important for lymphocyte homing are involved in lichen planus, including TNF-α, IL-1, IL-6, IL-8, and IFN-γ [12, 13]. These cytokines are derived from resident macrophages, lymphocytes, and keratinocytes. The chemokine RANTES (Regulated on Activation, Normal T-cell Expressed and Secreted) is thought to be important in oral lichen planus [14]. RANTES can induce mast cell degranulation, releasing pro-inflammatory mediators to induce an inflammatory reaction in lichen planus.
Lichen planus may have a genetic susceptibility factor as some patients have a positive family history of disease [15, 16]. HLA A-3 and HLA-A5 have been documented to occur at a higher prevalence in patients with lichen planus [17].
Lichen planus may be associated with viral infections, specifically hepatitis C virus (HCV) [18, 19]. In a meta-analysis, 16 % of patients with lichen planus had hepatitis C virus infection [19], suggesting a need for infectious workup in patients with widespread or unusual presentations of lichen planus. Of note, this association was primarily seen in adult patients and not in children with lichen planus, mitigating the need for screening in this population. Hepatitis C virus infection and lichen planus in the oral mucosa could be related to immunogenetic factors, such as the HLA-DR6 [20, 21]. Oral lichen planus occurs less frequently in children than in adults, possibly because adults more often undergo dental procedures that utilize dental amalgam, which is associated with oral lichen planus. Although the amalgam material itself does not cause lichen planus, the corrosion of amalgam and the continuous contact of corroded amalgam with the oral mucosa are associated with increased risk of oral lesions [22, 23].
4.2 Lichen Striatus
4.2.1 Clinical Features
Lichen striatus is an acquired asymptomatic lichenoid dermatosis, most frequently occurring in children between 4 months old and 15 years old, although many report median age to be closer to 2–3 years old [1]. There is no racial or gender predilection.
The physical exam is characterized by skin-colored to slightly erythematous, flat-topped 1–2 mm papules in blaschkoid distribution along the affected area (Fig. 4.4). Multiple bands may be seen with bilateral involvement described, although this is uncommon [1]. Skin changes are most often seen on the limbs but may occur on the face or trunk as well. It often progresses linearly with possible nail dystrophy if it involves the distal digits. As skin changes progress, lesions flatten and hypopigmentation becomes a more prominent feature. Vesicular variants have been reported [1]. Lichen striatus is characterized by spontaneous resolution of active dermatitis within 1 year, but residual hypopigmentation may persist for years.

Fig. 4.4
Lichen striatus presents as a Blaschkoid erythematous papular plaque extending distally from the right posterior shoulder down the arm of a boy
4.2.2 Histology
The diagnosis of lichen striatus requires careful clinicopathologic correlation. The epidermis is generally either unremarkable or displays focal spongiosis that some authors have described as occurring within the lower portions of the epidermis [24]. Parakeratosis is present in many cases. Acanthosis may be seen with elongation of the rete ridges [25]. An interface dermatitis is present, although often there is only a focal lymphocytic infiltrate of the basal layer leading to keratinocyte vacuolization and destruction (Figs. 4.5 and 4.6). Earlier lesions tend to have a more confluent and band-like infiltrate along the epidermal junction, and this diminishes in intensity with progressive age of the lesions. The dermis contains a superficial and deep perivascular infiltrate of lymphocytes and histiocytes. A similar infiltrate is seen around hair follicles and especially around eccrine coils [26]. Eosinophils and plasma cells are not part of the reactive process in most cases. While there are histologic features for lichen striatus as described, for the most part these are not specific and can be seen in a variety of lesions.
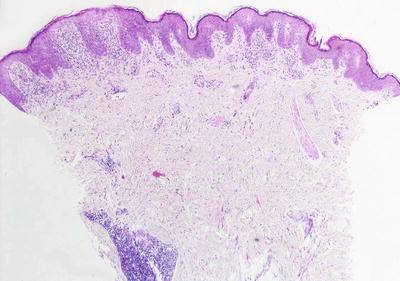
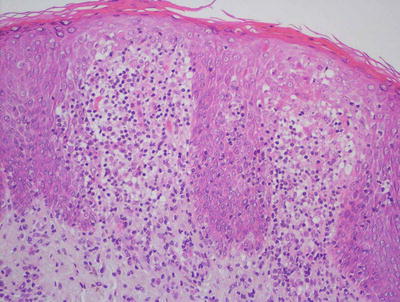
Fig. 4.5
A papillomatous epidermis with an underlying band-like infiltrate is characteristic of lichen striatus. A dense inflammatory infiltrate is also seen surrounding eccrine ducts
Fig. 4.6
An intense interface dermatitis with abundant exocytosis and focal spongiosis within an acanthotic epidermis is seen in some cases of lichen striatus
The histologic differential diagnosis includes lichen planus, lupus erythematosus, lichen aureus and arthropod bite reactions. Lichen planus may present the greatest diagnostic dilemma, especially the linear variant [27]. Early lesions of lichen striatus will demonstrate a similar band-like infiltrate along the epidermal junction; however, in lichen striatus, there is typically a deep component to the infiltrate and a noticeable peri-appendageal infiltrate that is not present in most cases of lichen planus. In addition, the focal hyperkeratosis that characterized lichen planus is not a common feature in lichen striatus, which often shows parakeratosis that is not seen in lichen planus. Lupus erythematosus presents a diagnostic challenge due to the interface reaction pattern superimpose upon the superficial and deep perivascular and peri-appendageal infiltrate similar to the pattern seen in classic lichen striatus. In most cases, this does not present a clinical diagnostic dilemma and can easily be resolved. The lack of epidermal atrophy, basement membrane thickening and dermal mucin in lichen striatus further helps in making this distinction. Lichen aureus shows a lichenoid inflammatory infiltrate similar to lichen striatus, but this is accompanied by extravasation of erythrocytes and the presence of papillary dermal hemosiderin, which are features not seen in lichen striatus. Arthropod bite reactions may have a similar inflammatory reaction pattern, but in most cases, eosinophils and neutrophils are also present within the infiltrate. In addition, while cutaneous appendages may be involved, there is no apparent predilection for them as typically seen in lichen striatus.
4.2.3 Pathogenesis
The pathogenesis of lichen striatus involves a combination of environmental and inherent genetic factors. The fact that lichen striatus occurs along the lines of Blaschko (Blaschkitis) suggests that a somatic mutation occurring during embryogenesis may be involved in its etiology [1, 28, 29]. Blaschko’s lines have an embryologic origin and correspond to the direction of growth of skin cells. Somatic mutations occurring during embryogenesis may produce an aberrant skin cell clone that is predisposed to give rise to lichen striatus [29]. This is further supported by reports of recurrent lesions in the same location [28]. Lichen striatus has also been considered to be a consequence of an acquired stimulus that induces the loss of immune tolerance resulting in a T-cell-mediated inflammatory reaction [29, 30]. It may also have an infectious etiology. Viruses have been implicated due to reports of multiple simultaneous cases in siblings, outbreaks among unrelated children in shared living quarters, and seasonal variation [31]. However, viral particles have not been identified in lesional skin [26]. Other possible precipitating factors include skin injury and trauma [32].
4.3 Lichen Aureus
4.3.1 Clinical Features
Lichen aureus is uncommon in children and more frequently presents in young adults [33]. No consistent sex predilection has been established, although some studies of pediatric populations suggest higher prevalence in females [1].
The appearance of lichen aureus is similar to that of a bruise, characterized by a gold to “rust-colored” patch or plaque of grouped lichenoid papules [1, 33] (Fig. 4.7). Lesions appear suddenly with slow expansion usually along the lower extremities, although any cutaneous surface can be affected. Multiple lesions may occur and occasionally coalesce into larger patches or plaques. Distribution is typically unilateral with involvement at the trunk and arms more frequently seen in children [33]. Patients may complain of mild pruritus, but lesions are most commonly asymptomatic. Lichen aureus has a self-limiting course, recurring in crops or persisting unchanged for years. Lesions occurring in children resolve more readily with an approximate duration of 2.5 to 3 years [1, 34].
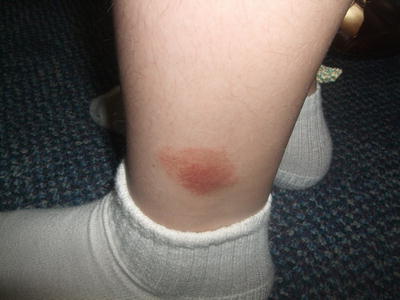
Fig. 4.7
Lichen aureus is characterized by a “rust-colored” circumscribed, non-blanching plaque on the lower extremity
4.3.2 Histology
Lichen aureus demonstrates a lichenoid inflammatory infiltrate along the epidermal junction, usually characterized by little destruction of the epidermal basal layer, similar to the histologic changes that have been described in adults with the same condition [33] (Figs. 4.8 and 4.9). The epidermis is unremarkable except for focal basal vacuolization and exocytosis of lymphocytes. Within the dermis, there is a dense lymphohistiocytic infiltrate usually confined to the papillary dermis [35]. The distinguishing diagnostic feature is the presence of erythrocyte extravasation with hemosiderin deposition, often accumulating within dermal macrophages. The gold brown pigment may be readily apparent on routine histologic sections, but can be better visualized on iron stains.
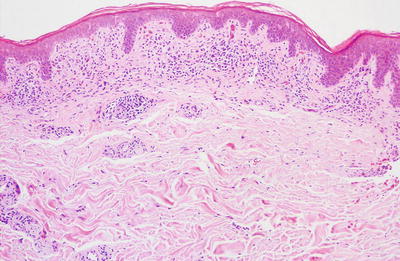
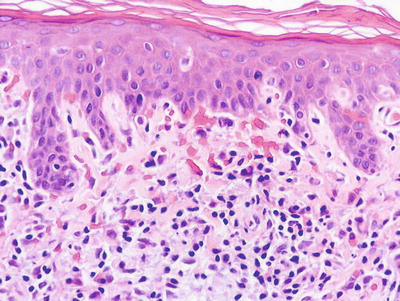
Fig. 4.8
Lichen aureus demonstrates a band-like infiltrate along the papillary dermis with focal hemorrhage
Fig. 4.9
Papillary dermal hemorrhage and a lymphocytic infiltrate without obvious vascular destruction are features of lichen aureus
The differential diagnosis includes other entities with dermal hemorrhage such as pityriasis lichenoides et varioliformis acuta (PLEVA) and pityriasis rosea . PLEVA is characterized by thick, usually confluent parakeratosis, which is not a feature of lichen aureus. In addition, the inflammatory infiltrate in PLEVA tends to extend deeper into the dermis than is usually seen in lichen aureus. Pityriasis rosea does not demonstrate the hemosiderin, has focal spongiosis, focal parakeratosis and a less intense inflammatory infiltrate than that seen in lichen aureus. Other types of pigmented purpuric eruptions may enter into the differential diagnosis, but the lichenoid pattern of inflammation is the defining feature of this subtype. The distinctions have no clinical or prognostic significance. Stasis dermatitis could be confused with lichen aureus in adults, but it would not be expected to enter the differential diagnosis in children. Leukocytoclastic vasculitis is often the major clinical differential diagnosis but is easily separated from lichen aureus based upon the findings of a neutrophilic infiltrate, vascular destruction, thrombosis and in more florid cases, epidermal necrosis. The major histologic differential diagnosis of clinical significance is mycosis fungoides . Rare cases of mycosis fungoides have been reported in children. Mycosis fungoides has more infiltration of the epidermis by lymphocytes, some of which may show cytologic atypia. Dying keratinocytes are not seen in most cases, and the infiltrate is rarely a confluent band within the papillary dermis. Dermal hemorrhage is unusual in mycosis fungoides . While clonality has been described in some cases of lichen aureus, most authors do not believe that it has the propensity for progression to mycosis fungoides [35, 36].
4.3.3 Pathogenesis
Several mechanisms of pathogenesis of lichen aureus have been suggested, including capillary fragility, benign chronic capillaritis, and possible exogenous factors such as infections, drugs, and contact allergens [37–39].
There are reports describing clonal T lymphocyte infiltrates in lichen aureus lesions with some suggestions of progression to mycosis fungoides [36, 40, 41]. However, malignant transformation to mycosis fungoides is not the typical course. A study showed no observed progression to mycosis fungoides in 23 patients with conventional lichen aureus [42]. It has been suggested that lichen aureus may be a type of dermatoses with clonal T cell expansion, in which the clone is kept under control by immune surveillance, but in a few patients, the clone may escape surveillance and become a dominant clone, predisposing the individual to cutaneous lymphoma.
4.4 Lichen Nitidus
4.4.1 Clinical Features
Lichen nitidus is an asymptomatic dermatosis of early childhood with most cases presenting before 10 years of age [1]. There is no gender or racial predilection. Physical exam is notable for generalized pinpoint (1–2 mm), monomorphic, skin-colored to slightly hypopigmented, flat-topped papules clustered at the trunk (abdomen, chest) and upper extremities (Fig. 4.10). Koebnerization is a key feature and is seen almost universally in affected persons. Fine scale or hyperkeratotic plug may overlie some papules. Generalized disease has been reported, presenting as fine hyperkeratosis with palmar involvement as well as ridging and pits of the nail plates [1]. Lichen nitidus eventually regresses, but it may persist for years.
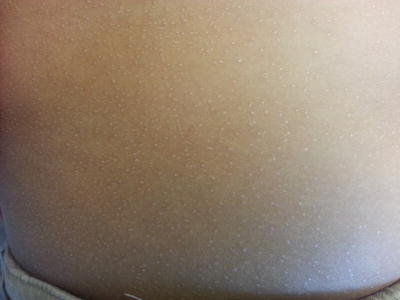
Fig. 4.10
Lichen nitidus frequently presents as monomorphic, flat-topped, skin-colored to hypopigmented papules with evidence of koebnerization on the abdomen of a child (photo courtesy of Audrey Chan, MD, Baylor College of Medicine, Houston, TX)
4.4.2 Histology
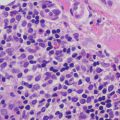
Lichen nitidus is characterized by what has been described as a “ball and claw” pattern . The epidermis demonstrates focal parakeratosis overlying areas of epidermal thinning. On either side of the thinned epidermis, there is a classic elongation of rete ridges with an inward turn, creating the “claw” pattern that surrounds the “ball” (Figs. 4.11 and 4.12). Within the papillary dermis, there is a focally dense inflammatory infiltrate that consists primarily of lymphocytes and histiocytes that comprises the “ball.” The infiltrate may extend into the lower portion of the epidermis, resulting in basal vacuolization and keratinocyte necrosis [43]. Extension around cutaneous appendages has been described in rare cases [44]. In some cases, there is an extensive histiocytic component that creates a granulomatous appearance, while in other cases the histiocytic infiltrate is relatively scant. Eosinophils and plasma cells are not prominent, although they may be seen in some cases [45]. The infiltrate is usually confined to the region subjacent to the flattened epidermis. Outside of this region, the dermis is relatively devoid of inflammation [43]. Rare cases of transepidermal elimination and perforation have been described [46, 47].
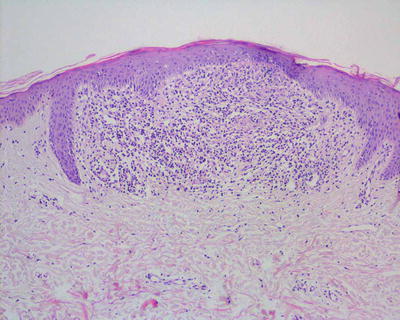
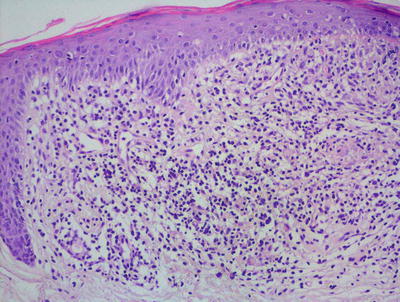
Fig. 4.11
Lichen nitidus demonstrates the classic “ball and claw” configuration with a small, circumscribed granulomatous inflammatory infiltrate enveloped by elongated rete ridges
Fig. 4.12
The inflammation in lichen nitidus consists of lymphocytes and abundant histiocytes largely within the dermis with some extension into the overlying epidermis
The histologic differential diagnosis includes lichen planus and granulomatous processes. Lichen planus has a more diffuse dermal infiltrate along the epidermal junction. The wedge-shaped hypergranulosis and altered shape of the rete ridges that characterize lichen planus are not present in lichen nitidus. In most cases, the histiocytic infiltrate in lichen nitidus is relatively scant, and infectious processes would not enter into the differential diagnosis. However, in rare cases with an abundance of histiocytes, a granulomatous appearance may suggest sarcoidosis, histoplasmosis and tuberculosis [48]. Special stains for microorganisms and clinical history will easily resolve this dilemma. Another differential consideration is lichen striatus, which has a more diffuse inflammatory infiltrate along the epidermal junction, and also has lymphocytes deeper within the dermis and around eccrine structures, which are features not associated with lichen nitidus.
4.4.3 Pathogenesis
Lichen nitidus is associated with immune dysfunction, particularly in cases of generalized disease [1]. It may be closely related to lichen planus. This is supported by the coexistence of both diseases in some patients, overlapping histomorphologic features, and similar infiltrates of CD4+ T lymphocytes [49, 50]. Lichen nitidus can occur in patients with atopic dermatitis, advanced HIV, and Crohn’s disease [51–53]. Some cases occurring in families have been reported [54, 55].
4.5 Lichenoid Drug Eruptions
4.5.1 Clinical Features
Incidence of lichenoid drug eruption in children is rare, since many of the causative agents are infrequently used in this patient population [1, 56]. The time of onset of skin changes in lichenoid drug eruption typically is longer than that seen in other drug reactions, with latency of approximately 12 months [56]. The associated skin lesions may mimic those of lichen planus, or may have a more eczematous appearance with larger discrete papules, more pronounced erythema, lichenification and scale [1]. Additionally, lichenoid drug eruption often spares the flexural wrist, genitals, and mucosal surfaces with rare reports of photodistribution. Skin eruption typically resolves after discontinuing the causative agent, although it may demonstrate slower resolution in comparison to other types of drug eruptions [56].
4.5.2 Histology
Drug eruptions manifest with many histologic patterns, one of which is a histologic mimic of lichen planus. In these cases, the epidermis may be slightly acanthotic with variable hyperkeratosis. Parakeratosis is present in some cases. The papillary dermis demonstrates a band-like inflammatory infiltrate that invades the lower portion of the epidermis, resulting in basal vacuolization, dying keratinocytes, and even small subepidermal blister formation (Figs. 4.13 and 4.14). In most cases, eosinophils and/or plasma cells will be present in addition to lymphocytes and histiocytes [1]. Their presence helps to make this distinction, along with a good clinical history. Further more, lichenoid drug eruptions often display involvement of the deeper dermis, which is not usually seen in lichen planus. Another differential diagnosis is an arthropod bite reaction. Arthropod bite reactions have a different clinical presentation from a lichenoid drug eruption . The presence of a punctum, dermal hemorrhage, and frequent eosinophils extending between reticular dermal collagen bundles all favor an insect bite reaction over a lichenoid drug eruption.
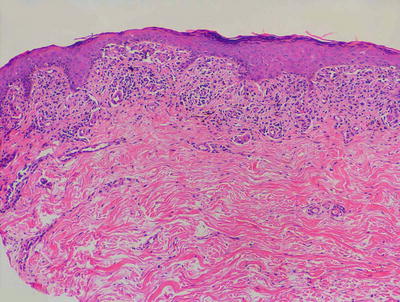
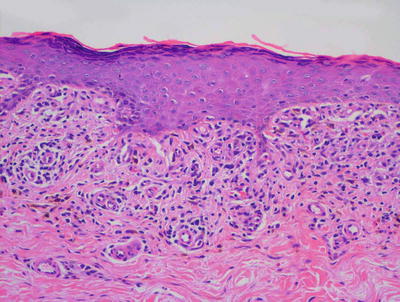
Fig. 4.13
Lichenoid drug eruption demonstrates a band-like infiltrate of lymphocytes in the papillary dermis with focal exocytosis into the epidermis. Hypergranulosis and rare dying keratinocytes may be present
Fig. 4.14
Hypergranulosis , a band-like lymphocytic infiltrate and pigment-laden macrophages are present in lichenoid drug eruption
4.5.3 Pathogenesis
Many clinically heterogeneous inflammatory dermatoses ranging from lupus erythematosus to fixed drug eruption share overlapping elements of a lichenoid inflammatory reaction [57]. Autoimmune attack by T lymphocytes in the epidermis is the primary pathological event in lichenoid dermatitis [57]. CD8+ T lymphocytes that are primed and activated by an infection, such as viruses, could evolve into long-lived effector memory T cells, and become effectors of epidermal damage seen in a lichenoid tissue reaction when they are exposed to stimuli, such as drugs or self-antigens. The severity of damages in the epidermis is dependent on the relative balance between the intensity and duration of T lymphocyte-mediated inflammatory reaction, as well as the capacity of epidermal cells to be protected from the immune response.
4.6 Lupus Erythematosus
4.6.1 Clinical Features
Juvenile systemic lupus erythematosus (JSLE) is a systemic autoimmune connective tissue disorder. It is estimated that approximately 10–20 % of patients with systemic lupus erythematosus develop signs and symptoms of disease during childhood and adolescence [58, 59]. Thirty-five percent of pediatric patients present between 5 and 10 years of age, and 5 % occur in children under 5 years of age. The overwhelming majority of cases are diagnosed in postpubertal children between the ages of 11 and 15 years old (60 %) with median age of onset of 12.1 years old [58, 60–62].
The incidence of JSLE is between 0.3 and 0.9 per 100,000 per year while prevalence ranges from 0.3 to 8.8 per 100,000 [58]. It is more common in females with a female to male ratio of 3–5 to 1. Children and adolescents often present with more acute and severe disease than those with adult onset lupus, including more significant organ involvement [59].
A variety of mucocutaneous manifestations are seen in JSLE (Figs. 4.15, 4.16 and 4.17). The lesions of acute cutaneous lupus erythematosus (ACLE) include localized and generalized lesions that are photosensitive. The most common localized lesion is a malar (“butterfly”) rash , characterized by a well-defined erythematous, edematous plaque over the nasal bridge (Fig. 4.15). In one study, the malar rash was reported in approximately 80 % of children with systemic lupus [60]. A more diffuse erythematous rash affecting non-sun exposed areas can also occur. Subacute cutaneous lupus (SCLE) is extremely rare. It is characterized by widespread annular and psoriasiform lesions.
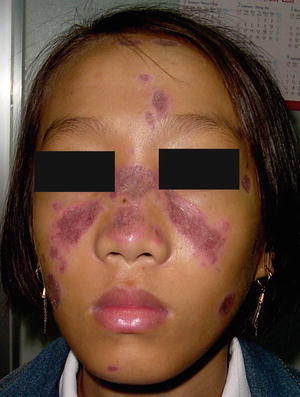
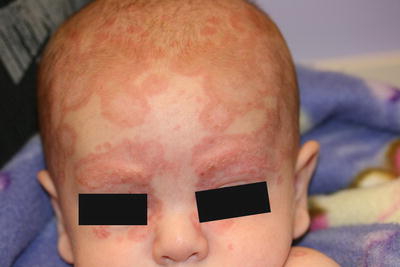
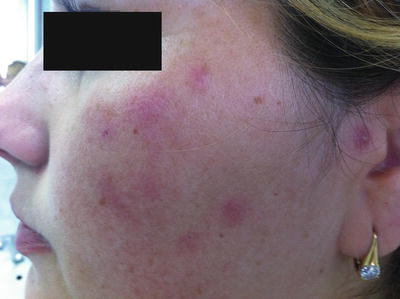
Fig. 4.15
Multiple violaceous atrophic plaques with an erythematous rim are on the face, most notably in a malar distribution across the nose and cheeks in a girl with lupus erythematosus (photo courtesy of Minh Hoang, MD, Ho Chi Minh City, Vietnam)
Fig. 4.16
Neonatal lupus erythematosus presents with annular scaled, erythematous thin plaques and patches scattered on the scalp and face an infant
Fig. 4.17
Tumid lupus erythematosus is characterized by smooth, indurated erythematous plaques on the head and neck with minimal epidermal change (photo courtesy of Irina Margaritescu, MD, Bucharest, Romania)
Discoid lesions of chronic cutaneous lupus (CCLE) are unusual in children under 10 years old. They are most common on the scalp, face and ears, and present as well-circumscribed, indurated, red-violaceous papules and plaques with adherent, keratotic scale or fine telangiectasia that cause atrophic scarring. Only approximately 25 % of children with discoid lupus go on to develop systemic disease [63]. Other forms of CCLE, such as lupus panniculitis and tumid lupus, are also rare in children (Fig. 4.17).
A variety of nonspecific skin lesions and changes can also be seen in JSLE . These include oral ulcers, non-scarring alopecia, photosensitivity, leukocytoclastic vasculitis, livedo reticularis, Raynaud’s phenomenon, and bullae, to name a few.
Management of JSLE is tailored to severity and presence of organ involvement. Systemic steroids and other immunosuppressants are mainstays of therapy for systemic disease. Aggressive photoprotection is of paramount importance. Disease prognosis varies widely depending on disease severity, patient compliance, and response to therapy.
Neonatal lupus is a special subtype of lupus erythematosus, and is a relatively rare disease that affects 1 in 10,000–20,000 newborns [64]. It is more commonly seen in females. Cutaneous manifestations are present in 50 % of cases with only 10 % of patients having coexistence of both cutaneous disease and complete atrioventricular block (the most severe variant) [64]. Approximately 25 % of patients have hepatobiliary or hematologic abnormalities with transient thrombocytopenia [62].
The skin eruption in neonatal lupus typically appears within the first few weeks of life, although congenital lesions have been reported. Patients most commonly present with erythematous annular plaques with predilection for the face and scalp (Fig. 4.16). Periocular erythema or “raccoon eyes ” is a frequently described clinical finding. Post-inflammatory dyschromia may occur, but lesions tend to resolve without scarring [64]. Almost all subtypes of neonatal lupus have a good prognosis with disease manifestations subsiding by about 1 year of life, which corresponds to the clearance of maternal autoantibodies. The one exception is disease associated with complete atrioventricular block, which is frequently more chronic and requires a pacemaker [64].
4.6.2 Histology
There are many clinical subtypes of lupus erythematosus , some of which can be differentiated on routine histologic sections. The most common subtype encountered is discoid lupus erythematosus and it demonstrates the most classic features of cutaneous involvement by lupus erythematosus. Histologic changes include orthokeratotic hyperkeratosis with follicular plugging and dilatation (Figs. 4.18 and 4.19). In well-developed lesions, the epidermis is atrophic, but in early lesions, the epidermis may have normal thickness. There is an interface dermatitis with scattered dying keratinocytes and lymphocytes extending into the lower portions of the epidermis (Figs. 4.20 and 4.21). In long-standing lesions, basement membrane thickening may be apparent on routine sections or on PAS stains. The dermis is characterized by a superficial and deep perivascular and peri-appendageal inflammatory infiltrate consisting primarily of lymphocytes. Scattered plasma cells are seen in some cases, but the presence of eosinophils is unusual. Dermal mucin may be present but usually in relatively small amounts. H&E sections may show mucin , which can be highlighted with colloidal iron or alcian blue stains.
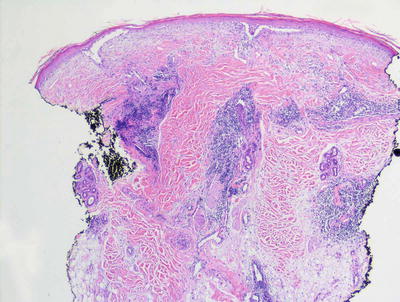
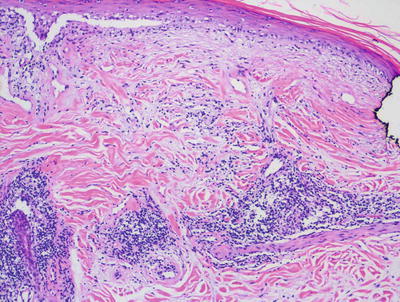
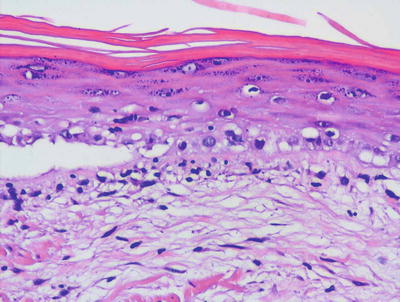
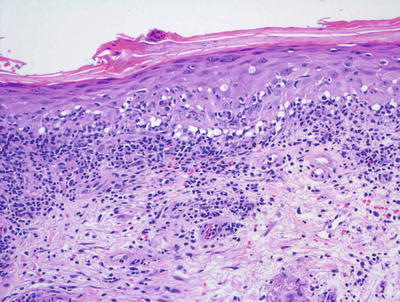
Fig. 4.18
Hyperkeratosis , atrophic epidermis with interface dermatitis, and a superficial and deep perivascular and peri-appendageal lymphocytic infiltrate characterize lupus erythematosus
Fig. 4.19
Interface dermatitis resulting in epidermal atrophy and a perivascular lymphocytic infiltrate along with dermal mucin are seen in lupus erythematosus
Fig. 4.20
Diffuse basal vacuolization is present in lupus erythematosus
Fig. 4.21
Interface dermatitis with scattered dying keratinocytes in lupus erythematosus
The histologic features seen in subacute cutaneous lupus erythematosus are identical to those described in discoid lesions. While some authors have attempted to distinguish these subtypes based upon biopsy findings [65], others have not been able to replicate the findings [66].
Systemic lupus erythematosus demonstrates similar histologic changes to those described in discoid lupus erythematosus . However, in most cases, the changes are paradoxically less intense. This generalization is not significant enough, however, to make the distinction based purely on biopsy results. Direct immunofluorescence stains and serologic studies are warranted to make a precise diagnosis.
Tumid lupus erythematosus is a less common variant of cutaneous lupus erythematosus in which the changes are restricted to the dermis and the epidermis remains histologically normal. Within the dermis the characteristic features of lupus erythematosus are seen with a superficial and deep perivascular and peri-appendageal infiltrate of lymphocytes and occasional plasma cells [67]. In tumid lupus, mucin deposition is often more pronounced than in other subtypes [68]. Tumid lupus is generally considered to be identical to what has previously been called reticular erythematous mucinosis [69].
Bullous lupus erythematosus is a rare subtype of lupus erythematosus that presents with clinically apparent blisters [70–73]. This is reflected in the histologic changes wherein a subepidermal blister is present. The underlying papillary dermis displays a pronounced infiltrate of neutrophils. The remainder of the dermis typically demonstrates the more classic findings of lupus erythematosus with a superficial and deep perivascular and peri-appendageal infiltrate.
Lupus profundus is primarily a panniculitis and is described in details in the chapter on panniculitides. Some cases may show changes of cutaneous lupus erythematosus, but in many cases the changes are restricted either entirely or largely to the subcutaneous fat.
Neonatal lupus erythematosus is a rare condition caused by the transplacental passage of maternal autoantibodies. In most cases, the clinical and histologic features resemble those seen in subacute cutaneous lupus erythematosus [74–77]. There is an interface dermatitis with vacuolar degeneration of basal keratinocytes . A superficial and deep perivascular and peri-appendageal lymphocytic infiltrate is present. Epidermal atrophy and melanophage deposition are seen in some cases. More recently, several cases of neonatal lupus erythematosus have been descried that display a neutrophilic dermal infiltrate [78, 79]. In rare cases, the neutrophilic infiltrate may be admixed with large histiocytoid cells that express CD68, CD33, and myeloperoxidase [80]. Vascular ectasia has also been described in some patients with neonatal lupus and a resemblance to cutis marmorata telangiectatica congenita has been reported [74, 81].
Direct immunofluorescence examination is helpful in establishing the diagnosis in some cases; however, the sensitivity varies greatly depending upon the subtype of lupus erythematosus. A granular staining pattern with antibodies directed against IgG, IgA, IgM, and C3 is found in most lesional biopsies taken from patients with discoid lupus erythematosus. The sensitivity is much lower for those with subacute lupus, tumid lupus, and lupus profundus. Non-lesional skin will show similar immunodeposits in patients with systemic lupus erythematosus and in some patients with subacute lupus, but it is otherwise usually negative.
The histologic differential diagnosis includes many entities, but the most commonly encountered ones are polymorphous light eruption, Jessner’s lymphocytic infiltrate, dermatomyositis, lymphoma, and cutaneous lymphoid hyperplasia. Polymorphous light eruptions do not demonstrate an interface dermatitis and peri-adnexal inflammation. Similarly, Jessner’s lymphocytic infiltrate would not be expected to show an interface process ; however, this entity is poorly defined and many authors believe some cases to be on the spectrum of cutaneous lupus erythematosus.
4.6.3 Pathogenesis
Lupus erythematosus is an autoimmune disease characterized by the production of self-antibodies to cell nuclear components. Genetic and experimental animal studies have provided important insights into toll-like receptor (TLR) and type I interferon (IFN) signaling in lupus erythematosus . An IFN-induced gene signature has been identified in children and adults with active systemic lupus erythematosus (SLE) [82–84]. There is a unique pattern of 14 upregulated gene targets of type I IFN, some of which are related to known autoantigens in SLE [82, 83, 85, 86]. Immune complexes of DNA and anti-double-stranded DNA antibodies that are normally found in the serum of patients with SLE can induce plasmacytoid dendritic cells (pDC) to secrete IFN-α [87]. pDC and IFN gene activation have been observed in skin lesions and nephritic glomeruli from SLE patients [88]. About 25 % of SLE patients have endogenous anti-IFN-α antibodies in the serum [89]. These patients also have reduced serum IFN levels as well as decreased IFN-pathway and disease activity. These findings indicate that inhibition of IFN activity may ameliorate disease condition in SLE. In fact, anti-IFN therapies are being developed for lupus [90]. Some of the therapies are humanized monoclonal antibodies to IFN-α and IFNα/β receptor. IFN-α blockade significantly reduces the IFN-gene signature in patients, although ultimately there have been no significant effects on disease activity thus far.
Experimental animal models of lupus erythematosus have shed important insights into the role of IFN and interleukin-1 pathways in the disease. IRAK1 (interleukin-1 receptor associated kinase-1) has been identified as a risk gene in the pathogenesis of SLE [91]. Deficiency of type I IFN receptor or IRAK1 in lupus-susceptible mice results in improvement in lupus disease phenotype [91, 92]. Moreover, chronic type I IFN production due to mutations in IFIH1 (a gene that encodes the cytoplasmic double stranded RNA sensor MDA5) results in aggressive disease in lupus-susceptible mice [93].
Dendritic cells are important in the pathogenesis of lupus erythematosus. In addition to its role in producing IFN, dendritic cells are necessary for B cell activation and tolerance [94, 95]. Lupus serum can induce dendritic cells to promote the differentiation of naive and memory B lymphocytes into IgG- and IgA-secreting plasma cells.
Neutrophils also play an important role in lupus erythematosus by contributing to the dysregulation of IFN in this disease. Anti-ribonucleoprotein antibodies found in SLE can stimulate neutrophils to release neutrophil extracellular traps (NETs) , which are composed of DNA and protein complexes [96]. NET can activate pDCs to increase levels of IFN-α in a DNA- and TLR-dependent manner. Another mechanism of neutrophil involvement is that neutrophils can undergo accelerated spontaneous apoptosis in vitro. Serum from lupus patients can induce neutrophil apoptosis and the release of neutrophil nuclear antigens, such as double stranded DNA, leading to the production of autoantibodies [97, 98].
There are familial cases of lupus erythematosus with pediatric onset that have a Mendelian pattern of inheritance. Genetic defects of the early complement components, namely homozygous C1q (90 % of cases), C4 (60 %), or C2 (30 %) deficiency, confer a high genetic risk of developing lupus [99]. DNA sequencing of affected siblings in a family with SLE identified a homozygous missense mutation in PRKCD gene, which encodes protein kinase C-δ (PKC-δ) [100]. Mutations in PKC-δ result in increased B lymphocyte survival and proliferation. Affected individuals have increased immature B lymphocytes and a developmental shift towards an immature phenotype of naive B lymphocytes. Experimental animal model with deficiency in PKC-δ develops SLE-like features, and has defects in the negative selection of self-reactive B cells [101, 102].
A rare autosomal recessive form of SLE with a fully penetrant pattern of Mendelian inheritance has been identified, in which there is a null mutation in the DNASE1L3 gene [103]. The onset of SLE related to DNASE1L3 occurs in children at 5 years of age. Additionally, a gene linked to a rare autosomal dominant form of SLE is heterozygous null allele in DNASE1, which encodes for a deoxyribonuclease enzyme [104]. These genetic findings raise a hypothesis that the inability to clear DNA materials released from apoptotic cells can trigger an autoimmune response, and the formation of anti-DNA antibodies with disease manifestations of lupus erythematosus.
4.7 Juvenile Dermatomyositis
4.7.1 Clinical Features
Juvenile dermatomyositis (JDM) accounts for more than 80 % of the cases of idiopathic inflammatory myopathies seen in children [105]. The annual incidence of disease is approximately 3 per million children per year in both the USA and the UK [106]. Girls are more commonly affected than boys with a mean age of onset of 7 years old, although a quarter will be diagnosed by 4 years old [106]. Non-Hispanic whites represent the overwhelming majority of those affected.
The vasculopathy of JDM affects the skin, skeletal muscles, gastrointestinal tract, lungs, kidneys, eyes, and heart [106]. The majority of children with dermatomyositis will have cutaneous findings at the time of diagnosis . The most frequently seen pathognomonic features are the heliotrope rash , Gottron’s papules and periungal changes of erythema, telangiectasia, hyperkeratosis, and fraying of the proximal nail fold. Sun-protected areas are frequently spared, suggesting photosensitivity .
The heliotrope rash is described as a violaceous to red erythematous eruption with discrete telangiectasia around the eyes and potential extension to the upper cheeks, forehead, temples, and ear. Similarly scaled dermatitis at the scalp, nape of the neck, extensor surfaces of the arms, shoulders, and upper trunk are qualified as the “shawl sign ”. Gottron’s papules are violaceous, flat-topped papules along the dorsal interphalangeal joints with subsequent evolution to poikiloderma (which may also be seen at other sites as a feature of more chronic disease) (Fig. 4.22).
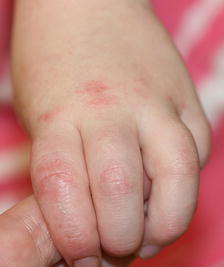
Fig. 4.22
Erythematous, scaled patches and thin plaques present on the dorsum of the hand and interphalangeal joints of a boy with dermatomyositis
Additional clinical features may include oral telangiectasia with concomitant erosions and ulcerations along the gums, buccal mucosa, and palate. Also present are palmar erythema and hyperkeratosis with fissuring (mechanic’s hands). Acquired lipodystrophy and associated metabolic derangements are seen in 10–40 % of children with long-standing and poorly controlled disease [105, 106]. Tender panniculitis also may occur at the arms, thigh, and buttocks. Lastly, firm, irregular nodules of cutaneous calcinosis that extrude whitish, chalky material can be seen in 30 % of pediatric cases, most commonly along pressure points or sites of trauma [106].
One-third of pediatric patients will enter complete remission within 3 years of disease onset, while the remainder will experience more persistent disease with polycyclic relapses [105]. Cutaneous ulceration may be seen in up to 10 % of patients and portends a more severe disease course. Overall, cutaneous disease typically responds well to immunosuppressive therapy, and children fare better than their adult counterparts with a lower associated mortality rate of less than 2 % [105].
4.7.2 Histology
Histologic findings in JDM are similar to those seen in lupus erythematosus. The epidermis is either normal or slightly atrophic with basal vacuolization (Figs. 4.23 and 4.24). Lymphocytes are present within the epidermis, papillary dermis and surrounding the vessels of the superficial vascular plexus. The inflammatory infiltrate does not usually extend deep into the reticular dermis, and does not involve cutaneous appendages. Occasional plasma cells may be encountered, but eosinophils are extremely uncommon in this condition. Dermal mucin is readily apparent on routine sections in most cases of dermatomyositis, but can be highlighted on colloidal iron or alcian blue stains [107–109]. Some authors have emphasized the finding of thickened blood vessels and sclerotic collagen in late stage lesions, which are features that may raise the question of scleroderma in some cases [110].
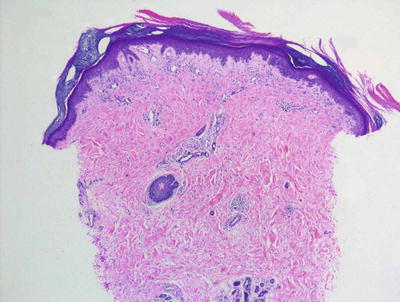
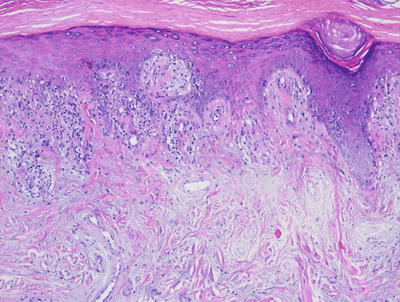
Fig. 4.23
Dermatomyositis demonstrates only a slight superficial interface dermatitis without deep or peri-appendageal inflammatory infiltrate
Fig. 4.24
Dermatomyositis has interface dermatitis with mucin present in the superficial dermis
Gottron’s papules have very similar histologic changes to those seen in biopsies taken from the poikilodermatous or erythematous lesions of dermatomyositis (Fig. 4.25). The major difference is that the epidermis is acanthotic and papillomatous with overlying hyperkeratosis rather than being atrophic [111]. An interface dermatitis with basal vacuolization, superficial lymphocytic infiltrate, and extensive dermal mucin characterizes the major histologic alterations in Gottron’s papules.
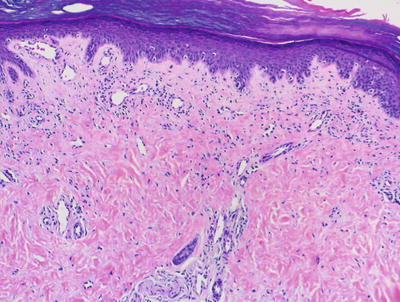
Fig. 4.25
A Gottron’s papule has the same histologic features as dermatomyositis affecting other areas of the skin but is present on acral skin
A consequence of long-standing dermatomyositis in children is the presence of diffuse dermal calcinosis, known as calcinosis universalis . Histologic changes may or may not show features consistent with active lesions of dermatomyositis . In most cases, scattered lymphocytes and plasma cells are present along with increased numbers of fibroblasts. Elastic tissue fibers within the reticular dermis demonstrate a granular blue appearance, consistent with calcium deposition [112, 113].
4.7.3 Pathogenesis
The pathogenesis of juvenile dermatomyositis is believed to involve a combination of environmental triggers and dysfunction of the adaptive immune system in genetically susceptible individuals. Certain HLA alleles that may be important in JDM susceptibility, including HLA-DQA1*0501, HLA-DQA*0301, HLA-DRB1*0301, and HLA-B8, have been found more frequently in patients with JDM [105]. Some studies suggest that HLA-dependent chimeric cells play a direct role in disease pathogenesis, and that the mother’s HLA genotype facilitates the transfer and persistence of maternal cells in the fetal circulation [114].
The immune system contributes to the pathogenesis of JDM. Myositis-associated antibodies and myositis-specific autoantibodies (MSAs) have been identified in about 40 % of JDM patients [115]. These autoantibodies are directed against cytoplasmic and nuclear components of myocytes. Vasculitis and vessel injury occur in JDM through activated complement components that induce cytokine release and the formation of the membrane attack complex [105, 115, 116]. In children with dermatomyositis, MHC-I heavy chain and β2-microglobulin are overexpressed in affected muscle tissues [117]. Type 1 interferons (IFNα and IFNβ) and tumor necrosis factor-α (TNF-α) play a central role in dermatomyositis by upregulating MHC class I in muscle fibers and causing activation of the endoplasmic reticulum stress response, leading to muscle injury and resulting myositis [117–119]. Dermatomyositis in children and adults seem to correlate with the type 1 IFN gene signature [120]. In addition, IL-6 production and type 1 IFN gene signature are potential biomarkers for disease activity in dermatomyositis [120].
An abnormal response to a foreign antigen that is homologous to a self-antigen (molecular mimicry) may be another mechanism for the aberrant immune response seen in JDM . For example, the amino acid sequence of the M5 protein of group A β-hemolytic streptococci is homologous to human skeletal myosin heavy chain [121]. Moreover, myosin heavy chain epitope may be a target autoantigen in JDM with cytotoxic T lymphocytes contributing to muscle fiber damage. Other possible triggers include infectious agents (such as coxsackievirus, toxoplasma, echovirus, and parvovirus), sun exposure, medications, and vaccines [105, 122].
4.8 Erythema Multiforme and Stevens–Johnson Syndrome/Toxic Epidermal Necrolysis
4.8.1 Clinical Features
Erythema multiforme is relatively rare in children, accounting for 20 % of reported cases [123]. It is slightly more common in boys. Erythema multiforme typically is thought to represent a reactive process to an exogenous stimulus, most commonly drugs (such as penicillins) or infection (such as herpes simplex virus, mycoplasma pneumoniae, Epstein B virus, and group A streptococcus) [123, 124]. In pediatric cases, the most common cause is infection, and mycoplasma pneumoniae and herpes simplex virus are most frequently associated with disease onset [125].
“Classic” erythema multiforme is described as the acute onset of symmetrically distributed, dusky-colored, targetoid, erythematous macules and papules with acral accentuation, including the palms and soles (Fig. 4.26). Central vesiculation and erosions may be seen. Mucosal involvement may occur as well (coined by some as erythema multiforme major). Skin changes typically resolve within 2 weeks, but secondary dyschromia and scarring may persist for longer periods of time. Relapses do occur, especially in the setting of recurrent mucocutaneous herpes simplex virus infection [126].
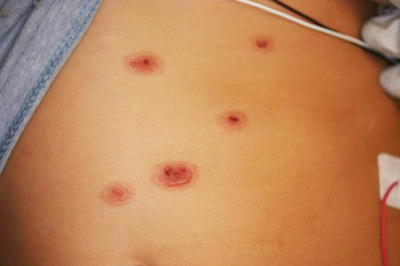
Fig. 4.26
Erythema multiforme shows targetoid erythematous papules and plaques with central vesiculation
Stevens–Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) is a delayed-type hypersensitivity reaction that causes widespread apoptosis of keratinocytes. In contrast to erythema multiforme, SJS/TEN is more commonly caused by drug exposure [125]. The main causative agents include anti-infective sulfonamides and anticonvulsants, specifically carbamazepine, followed by penicillins and nonsteroidal anti-inflammatory drugs [125, 127]. Mycoplasma pneumoniae is the leading infectious cause of disease seen in children, although cases following infections by viruses (such as coxsackievirus, influenza virus, Epstein–Barr virus, human herpes virus 6 and 7, cytomegalovirus, and parvovirus), bacteria (streptococcus β-haemolyticum group A), mycobacterium, and rickettsia have been described as well [128].
When skin detachment is limited to less than 10 % of body surface area involvement, patient is said to have SJS, while patient with more than 30 % of body surface area involvement is qualified as TEN. The skin exam of SJS/TEN is characterized by dusky-colored, erythematous macules and papules with eventual evolution to vesicles and bullae that coalesce into large areas of widespread necrotic desquamation (Figs. 4.27 and 4.28). Pain is a prominent feature as well as a positive Nikolsky sign (bulla formation when the skin is gently rubbed). The cutaneous presentation may be preceded by fever, malaise, ocular symptoms, and dysphagia [126, 128]. Almost all patients have involvement of at least two mucosal surfaces with similar vesicles, erosions, and overlying hemorrhagic crust. Children appear to have a better prognosis than adults when the offending drug is eliminated, or the underlying infection is treated [128]. Morbidity and mortality rates appear to improve with admission to a specialized burn unit or a pediatric intensive care unit. Long-term sequelae may include dyschromia, scarring (with hair loss or anonychia), mucosal strictures, and impaired vision.
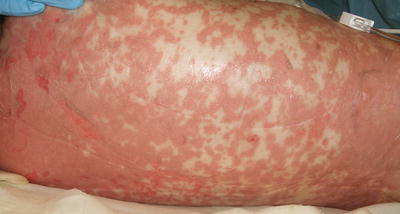
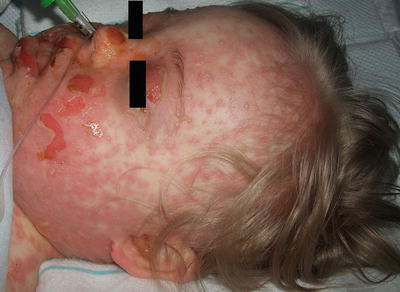
Fig. 4.27
Toxic epidermal necrolysis is characterized by dusky-colored, erythematous macules, papules and bullae with widespread necrotic desquamation
Fig. 4.28
Toxic epidermal necrolysis invariably involves mucosal surfaces with hemorrhagic crust overlying erosions at the mouth, cheeks, nose and eye lids
4.8.2 Histology
The histologic changes in erythema multiforme, Stevens–Johnson syndrome, and toxic epidermal necrolysis are quite similar and are described together. The stratum corneum is unremarkable. Specifically, except in old lesions, parakeratosis is not an expected finding, although mucosal biopsies in patients with Stevens–Johnson syndrome may demonstrate site-specific parakeratosis. This is important in distinguishing erythema multiforme from other inflammatory dermatoses with an interface pattern of inflammation. The epidermis has normal thickness, and it is characterized by varying amounts of basal vacuolization, keratinocyte necrosis, and lymphocytic exocytosis into the epidermis (Figs. 4.29 and 4.30). In TEN, full thickness epidermal necrosis is a common finding, often resulting in a subepidermal blister. In these cases, there is paradoxically a relatively scant inflammatory infiltrate. In the minor form of the disease, lymphocytes are present within the epidermis, along the dermal-epidermal junction and surrounding the superficial vessels [129]. In some cases, eosinophils may be present, but in many cases they are absent or inconspicuous. It has been suggested that there is preferential keratinocyte necrosis within acrosyringia in drug-induced erythema multiforme, likely due to accumulation of toxic levels of the drug within these structures [130]. A macrophage predominant pattern of exocytosis has been observed in some cases of erythema multiforme [131, 132]. An overlap syndrome with lupus erythematosus has been described (Rowell syndrome ) that demonstrates histologic features of both entities [133].
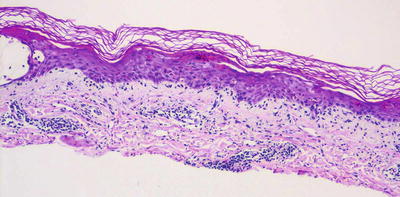
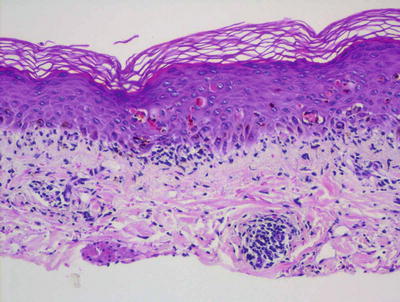
Fig. 4.29
Erythema multiforme is characterized by interface dermatitis and multiple dying keratinocytes, frequently giving rise to blisters
Fig. 4.30
Orthokeratotic keratin, exocytosis of lymphocytes into the epidermis and dying keratinocytes are the hallmark features of erythema multiforme
The differential diagnosis includes lupus erythematosus, dermatomyositis, graft vs. host disease, and pityriasis lichenoides. Lupus erythematosus can be distinguished based upon the presence of hyperkeratosis, a deep and peri-appendageal inflammatory infiltrate and dermal mucin, which are features not seen in erythema multiforme. Dermatomyositis similarly has abundant dermal mucin not seen in erythema multiforme. Graft vs. host disease can be very difficult to distinguish from erythema multiforme. The presence of eosinophils and a predilection for acrosyringeal necrosis might favor erythema multiforme, but often the clinical history is the best or only way to make this distinction. Pityriasis lichenoides demonstrates parakeratosis and hemorrhage, which are features not seen in erythema multiforme. TEN may be clinically confused with staphylococcal scalded skin syndrome. However, TEN results in a subepidermal blister, while a very superficial blister within the stratum corneum or uppermost portion of the granular layer characterizes staphylococcal scalded skin syndrome.
4.8.3 Pathogenesis
Erythema multiforme is a delayed-type hypersensitivity reaction that results in widespread apoptosis of epidermal keratinocytes. The pathogenesis is multifactorial and includes genetic and immune predispositions together with an inciting event, such as infection or medications [134]. In children, infectious causes that can induce erythema multiforme include bacteria (Mycoplasma pneumoniae, streptococcus, and rickettsiae) and viruses (herpesvirus and parvovirus) [128]. In most cases, the precipitating event is infection with herpes simplex virus (HSV) being involved in 70–80 % of cases [134–136]. Drugs , particularly sulfonamides, nonsteroidal anti-inflammatory drugs (NSAIDs) and anticonvulsants, are other key triggering factors. Although less common, the disease can also be associated with malignancies, autoimmune diseases, radiation, and food additives [134, 137].
Genetic factors, particularly human leukocyte antigens (HLAs) , predispose patients to develop erythema multiforme in response to stimuli. HLA-DQw3 has been found to be associated with HSV-associated erythema multiforme and may be a helpful marker to distinguish this entity from other erythema multiforme-like disorders [138]. Recurrent disease has been found to be associated with a number of HLAs, including HLA-B15 (B62), HLA-B35, HLA-A33, HLA-DR53, and HLADQB1*0301 [139, 140].
Studies of HSV-associated erythema multiforme have provided some insights into the mechanism of disease. Disease development starts with HSV infection of skin keratinocytes and mononuclear cells, such as macrophages and CD34+ Langerhans cell progenitors [141, 142]. Mononuclear cells carry HSV antigens (such as viral DNA fragments) to distant sites, leading to the recruitment of HSV-specific CD4+ type 1 T helper cells (Th1) [141, 143]. Th1 cells respond to viral antigens by producing interferon-γ (IFN-γ), which induces a robust immunologic response, causing epidermal damage. T-lymphocyte-mediated immune reaction results in cytotoxic immunological attacks on keratinocytes that express foreign viral antigens.
In drug-induced erythema multiforme, reactive drug metabolites trigger the disease process. Tumor necrosis factor-α (TNF-α), perforin, and granzyme B as well as cytotoxic CD8+ T lymphocytes cause keratinocyte apoptosis and epidermal damage [140]. Antibody-mediated immune response may also be involved in the disease.
Infection, vaccination, systemic diseases and drugs all have been implicated as the causes of Stevens–Johnson syndrome and toxic epidermal necrolysis. Among these potential sources, drugs are the most common inciting agents with antibiotics identified as the most frequent cause, followed by analgesics, anticonvulsants, and (NSAIDs) [144, 145]. The development of SJS/TEN is significantly higher in patients with immunosuppression, such as those with human immunodeficiency virus (HIV) infection and bone marrow transplantation [146, 147]. Mycoplasma pneumoniae leads to SJS predominantly in children and adolescents, the age group in which infections by this agent is most frequently seen [128, 148].
There is a strong genetic association between SJS/TEN and HLA allotypes, particularly in the Han Chinese population. In particular, HLA-B*1502 and HLA-B*5801 are associated with adverse drug reactions to carbamazepine and allopurinol [149, 150]. These findings are significant in that the FDA recommends genotyping all Asian patients for these HLAs before administration of drugs. These findings, however, have not been found in white Caucasian population [151]. An association between HLA-B*5801 and allopurinol-induced SJS/TEN has been found in the Japanese population [152]. Other HLA allotypes associated with adverse drug reactions have emerged, including HLA-A*3101 and HLA-B*1511 with carbamazepine , HLA-B*1502 with phenytoin, HLA-B*5801 with allopurinol, HLA-B*38 with sulfamethoxazole, and HLA-B*73 with NSAIDs [153]. Thus, an accurate knowledge of the risks associated with various HLAs would allow drugs to be tailored to an individual’s genetic risk.
A major pathological finding in SJS/TEN is widespread keratinocyte apoptosis. T lymphocytes are involved in epidermal destruction in this disease. This process involves Fas–Fas ligand interaction, CD8+ cytotoxic T lymphocytes and natural killer-cells, and TNF-α and IFN-γ [154, 155]. Keratinocytes undergo apoptosis mediated by Fas-Fas ligand [156, 157]. Keratinocytes in lesional skin of patients with TEN have high levels of Fas and FasL that interact with Fas and FasL on adjacent keratinocytes. Skin-infiltrating lymphocytes have also been found to express Fas ligand. Interaction of Fas and FasL leads to the activation of caspase enzymes and cell apoptosis [158]. CD8+ T lymphocytes and natural killer cells are present in high number in blister fluid of patients with TEN. These cells express granzyme B and perforin, and can trigger keratinocyte apoptosis by the perforin, granzyme B, and granulysin pathway [155]. Drug-specific CD8+ T lymphocyte clones have been found in the skin of patients with drug eruptions which may serve as a mediator of apoptosis in TEN [159, 160]. Fluid from blisters in TEN patients has high concentrations of IFN-γ, TNF-α, and interleukins 6, 13, and 18 [161]. These cytokines can increase the expression of Fas ligand on keratinocytes, upregulate adhesion molecules and induce T lymphocyte recruitment to the skin, thus further enhancing keratinocyte apoptosis. The discovery of the roles of Fas-Fas ligand and CD8+ cytotoxic T lymphocytes in SJS/TEN has led to improved therapy for this disorder, including IVIG containing anti-Fas antibodies that can block Fas–FasL interactions, and cyclosporine which inhibits cytotoxic T lymphocytes [154].
4.9 Phototoxic Dermatitis
4.9.1 Clinical Features
Phototoxic dermatitis is a non-immunologic skin reaction occurring minutes to hours after concomitant exposure to a photosensitizing substance and UV radiation. Reaction is dose dependent and universal in any person exposed to the substance in high enough quantities to elicit the photo-induced skin response [162].
Phytophotodermatitis (plant-induced photosensitivity) is the most common variant of phototoxic dermatitis seen in children, with plant-derived furocomarin compounds serving as the sensitizing agent. These naturally occurring psoralens may be found in a variety of plants, although the most frequently implicated families are Umbelliferae (celery, parsley, parsnip), Rutaceae (lemons, limes), and Moraceae (figs) [163]. Skin changes may mimic a sunburn with redness and occasional edema or appear in geometric patches and plaques with or without vesiculation. Pain may be reported at the site while pruritus occurs less frequently [163]. The acute dermatitis eventually evolves to hyperpigmentation and desquamation at the affected area.
4.9.2 Histology
Histologic sections of a phototoxic dermatitis show an epidermis of normal thickness with overlying orthokeratotic keratin. Within the epidermis, scant spongiosis is present along with scattered dying keratinocytes (Figs. 4.31 and 4.32). There is a mild lymphohistiocytic inflammatory infiltrate. Lymphocytes demonstrate exocytosis into the epidermis and are otherwise largely confined to the papillary dermis and around the superficial vessels. Eosinophils are infrequently seen in this type of drug eruption [164–166].
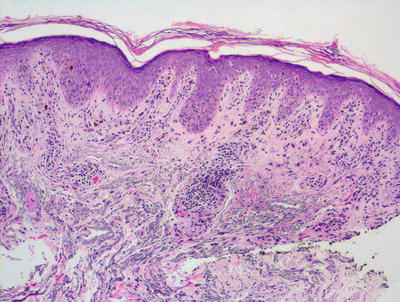
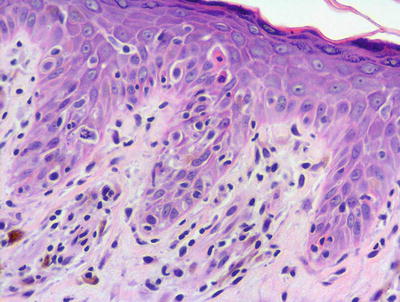
Fig. 4.31
Interface dermatitis with lymphocyte exocytosis and scattered dying keratinocytes characterize a phototoxic dermatitis
Fig. 4.32
Dying keratinocytes adjacent to intraepidermal lymphocytes are features of phototoxic dermatitis
The histologic differential diagnosis includes graft vs. host disease , erythema multiforme and other interface dermatoses such as lupus erythematosus , dermatomyositis , and pityriasis lichenoides . A polymorphous light eruption might also enter the differential diagnosis. Graft vs. host disease and erythema multiforme might only be differentiated from phototoxic dermatitis based upon clinical history and presentation, as the histologic changes are quite similar. Lupus erythematosus shows a deep and peri-appendageal infiltrate, and dermatomyositis has dermal mucin not seen in a phototoxic drug eruption. The parakeratosis that is common in pityriasis lichenoides would not be an expected finding in phototoxic drug eruptions. Polymorphous light eruption does not feature dying keratinocytes, and the papillary dermal edema and reticular dermal inflammatory infiltrate in polymorphous light eruption are not seen in phototoxic drug eruptions . A photoallergic drug eruption demonstrates abundant spongiosis and a significant eosinophilic component in the inflammatory infiltrate.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


