Group
PRR
PAMPs and DAMPs
Source of PAMPs and DAMPs
TLRs
TLR1/2
Triacyl lipopeptides
Bacteria
TLR2
Peptidoglycan
Gram (+) bacteria, e.g., S. aureus
Glycolipids
Mycobacteria
Phosphilipomannan
C. albicans
TLR3
dsRNA
dsRNA viruses
TLR4
Lipopolysaccharide
Gram (−) bacteria
TLR5
Flagellin
Flagellated bacteria, e.g. Salmonella typhimurium
TLR6/2
Diacyl lipopeptides
Mycoplasma
TLR7 and TLR8
ssRNA
Viruses, e.g., influenza, HIV-1
TLR9
Unmethylated CpG DNA
Bacteria
NLRs
NOD1
iE-DAP
Bacteria, e.g., H. pylori, Shigella flexneri, Listeria monocytogenes, E. coli, P. aeruginosa
NOD2
MDP
Bacteria, e.g., S. aureus, S. pneumonia, Shigella fexneri, Listeria monocytogenes, M. tuberculosis
NLRP1
Anthrax lethal toxin
Bacillus anthracis
NLRP3
MDP, DNA, RNA, toxins
Bacteria, e.g., L. monocytogenes, S. aureus
ATP, MSU, cholesterol
Endogenous
Silica, asbestos, alum
exogenous
NLRC4
Flagellin
Flagellated bacteria, e.g., S. tyhimurum, L. pneumonia, P. aeruginosa, S. flexneri, L. monocytogenes
AIM2
AIM2
dsDNA
Francisella tularensis, L. monocytogenes, vaccinia virus
RLRs
RIG-I
Short dsRNA, ssRNA
Viruses, e.g., HCV, VSV, reovirus
MDA5
Long dsRNA
Viruses, e.g., picornavirus, vaccinia virus
CLRs
Dectin-1
β-glucan
Fungi, e.g., C. albicans, Mycobacteria
Dectin-2
High mannose, α-mannans
C. albicans, C. neoformans, S. cerevisiae, M. tuberculosis
Innate immunity provides the first line of host defense, which can immediately respond to pathogens. Innate immunity was previously considered as a physical barrier provided by mucous membranes and ciliary action in the lungs and nasal passages, and the stratum corneum and keratinocytes’ tight junctions in the skin, which do not require specific recognition of microbes. Over the past two decades, however, there has been rapid progress in our understanding of immune responses to microbial components, which are evolutionally conserved between species. Currently, chemical and cellular barriers that specifically recognize microbes are regarded as the main component of innate immunity; chemical barriers include specialized soluble molecules that possess antimicrobial activity, whereas cellular barriers are comprised of an array of cells with sensitive receptors that detect microbial components.
Interestingly, the fundamental mechanisms of innate immunity and its basic intracellular signaling are conserved between plants and animals. In plants, for example, the innate-immunity–mediated necrotic cell death of infected cells and the subsequent release of danger signals in the local site can effectively block the proliferation and spreading of pathogens [2], whereas activation of mammalian innate immunity can cause lytic cell death, which can lead to efficient clearance of intracellular bacteria [3].
16.2 Pattern Recognition Receptors
Innate immunity senses the presence of a pathogen by recognizing molecules typical to a microbe but not shared by host cells, termed pathogen-associated molecular patterns (PAMPs). The major pattern recognition receptors (PRRs) of the innate immune system are Toll-like receptors (TLRs) , nucleotide-binding oligomerization domain (NOD), leucine-rich repeat (LRR) containing or Nod-like receptors (NLRs) , retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), and C-type lectin receptors (CLRs) [4]. Recognition of PAMPs by these receptors initiates several different mechanisms, such as the release of type I interferon and inflammatory cytokines, to eliminate the pathogen.
An array of PRRs are present on immune cells resident in skin, such as epidermal Langerhans cells (LCs), dermal dendritic cells (DCs), and mast cells, as well as nonimmune cells, such as keratinocytes, melanocytes, vascular endothelial cells, fibroblasts, and adipocytes. This wide distribution of PRRs in nonimmune cells in the skin is suitable for immediate self-defense response because skin is continuously exposed to microorganisms and also a target of an array of viral, bacterial, and fungal microbes, such as HPV, Streptococcus aureus, and Candida albicans, respectively. Therefore, the capacity of nonimmune cells, such as keratinocytes, to induce an immediate innate antimicrobial response is crucial.
TLRs recognize pathogens at the cell surface (TLRs 1, 2, 4–6, 10) or within the endosome (TLRs 3, 7–9), CLRs recognize fungal components at the cell surface, and NLRs and RLRs act as intracellular surveillance molecules [4, 5]. To date, 10 functional human TLRs [6] and 23 members of the human NLR protein family have been reported [7]. The RLR family contains retinoic-acid–inducible gene I (RIG-I) and melanoma-differentiation–associated gene 5 (MDA5), which sense cytosolic double-stranded RNA (dsRNA). The CLR family has a central role in immunity to fungal pathogens.
16.3 Toll-Like Receptors
Each TLR recognizes a distinct PAMP by forming a homo- or heterodimer. TLR2 can form a heterodimer with either TLR1 or TLR6 to recognize bacterial tri- or diacyl lipopeptides (LP), respectively [8], and TLR2 homodimer recognizes peptidoglycan (PGN) (Fig. 16.1). The other representative TLR-targeted molecules are dsRNA, which appears during the replication cycle of most viruses and is recognized by TLR3; lipopolysaccharide (LPS) from gram-negative bacteria, recognized by TLR4 along with CD14; bacterial flagellin, recognized by TLR5; viral single-stranded RNA (ssRNA) by TLR 7/8; and unmethylated CpG motifs, which are abundant in microbial DNA, recognized by TLR9 [8].
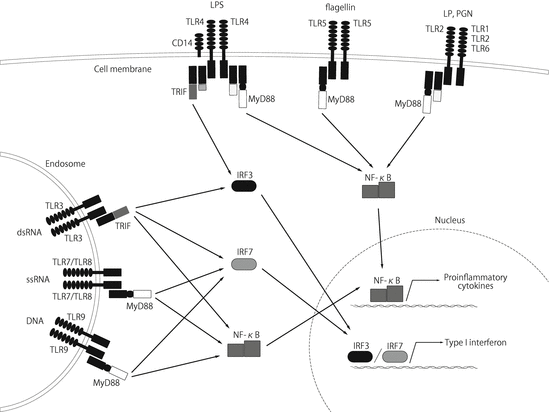
Fig. 16.1
TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10 are located in the plasma membrane and recognize mainly bacterial PAMPs, whereas TLR3, TLR7, TLR8, and TLR9 are located in the endosomal membrane and recognize mainly viral nucleic acids, such as dsRNA and ssRNA. The TLR signaling pathways are channeled through the adapter molecules MyD88 and/or TRIF. These adapter molecules activate NF-κB, resulting in the transcription of pro-inflammatory cytokines, whereas activation of endosomal TLRs and TLR4 leads to activation of IRF3 and IRF7, resulting in the production of type I interferon
All TLRs are type I transmembrane receptors that have ectodomains containing LRR that are involved in recognition of PAMPs, and transmembrane domains and intracellular Toll/interleukin-1 receptor (TIR) domains are required for downstream signal transduction. Once a TLR is activated, the TIR domain of the TLR interacts with an adapter molecule, such as MyD88 (myeloid differentiation factor-88) and TRIF (Toll/interleukin 1 receptor domain-containing adaptor-inducing interferon β) [4]. MyD88 is used by all TLRs except TLR3, and activates the transcription factor NF-κB and mitogen-activated protein kinases (MAPKs). The MyD88-dependent pathway leads to the production of inflammatory cytokines such as TNF-α, IL-1, IL-6, and IL-12, chemokines including IL-8 and MIP2, costimulatory molecules such as CD80 and CD86, and adhesion molecules such as ICAM-1. TLR3 and TLR4 trigger the MyD88-independent TRIF pathway that leads to activation of the transcription factors interferon regulatory factor 3 and 7 (IRF3/7) and NF-κB, which promotes type I interferon (interferon (IFN)-α/β) and inflammatory cytokine expression [4].
In human skin, TLRs are expressed in different cell types, from the epidermis to adipose tissue, with great variation in expression level and functionality. For instance, keratinocytes express functional TLRs 1–3 and 5–6 [9], and play a dynamic role in host defense beyond their role as a physical barrier. There is some controversy about the expression of TLR4 [10, 11], TLR7, and TLR9 [9, 12] in keratinocytes. Activation of TLR2 on keratinocytes by S. aureus results in IL-8 production, whereas activation of TLR3 by dsRNA leads to type I IFN production [13]. Epidermal LCs express all TLRs, particularly TLRs 1–3, 5, 6, and 10 [14], and produce IL-6, IL-8, and TNF-α after TLR2/3 stimulation [14]. Other cells, such as dermal monocytes/macrophages, dermal DCs, T and B cells, and mast cells, vascular endothelial cells, and skin stromal cells such as fibroblasts and adipocytes are known to express TLRs [15].
16.4 NLRs and the Inflammasome
The NLRs are all located in the cytoplasm and sense foreign or endogenous PAMPs or danger-associated molecular patterns (DAMPs), many of which are released from dying cells and following tissue injury, through an incompletely understood mechanism [16]. NLRs contain a centrally located NOD and an LRR domain near their carboxyl terminus. The LRR domain of NLRs is thought to play a role in the recognition of PAMPs and DAMPs, just as the LRR domain of TLRs does. The NOD is thought to be involved in the induction of conformational changes and self-oligomerization necessary for NLR function [16]. The NLRs are considered to be a very ancient family of innate immune receptors because the plant resistance (R) proteins responsible for defense against plant pathogens are NLR homologues. NLRs are grouped into subfamilies according to their additional domains. Typical additional domains present in NLRs are the caspase activation and recruitment domains (CARDs) and the pyrin domains (PYDs). These domains are involved in protein interactions, allowing for the recruitment of downstream effector molecules.
Widely studied members of the NLR family are NOD1 and NOD2 . These receptors recognize components of bacterial cell walls; specifically, NOD1 recognizes γ-D-glutamyl-meso-diaminopimelic acid (iE-DAP) [17] and NOD2 senses muramyl dipeptide (MDP) [18, 19]. Recognition of microbial products by NOD1/2 activates receptor-interacting serine-threonine kinase 2 (RIP2) via cellular inhibitors of apoptosis 1 and 2 (cIAP1 and 2), subsequently leading to ubiquitination of NF-κB essential modulator (NEMO) and activation of the pro-inflammatory NF-κB pathway [20]. The expression of NOD1/2 was first described in antigen-presenting cells such as monocytes/macrophages, but recent studies have revealed the functional expression of NOD1/2 in human keratinocytes [21, 22].
Another well-studied group of NLR family genes is NLRP1, NLRP3, and NLRC4, which form a multiprotein complex, termed the inflammasome. A variety of ligands can activate these receptors, particularly NLRP3 (Table 16.1). In addition to PAMPs such as ssRNA R837, the pore-forming toxin hemolysin derived from S. aureus and nigericin, many DAMPs , such as environmental pollutant crystals including silica and asbestos, vaccine adjuvants such as aluminum salt (alum) and endogenous metabolic stresses such as high glucose, cholesterol, β-amyloid, monosodium urate crystals (MSU), and extracellular ATP, can activate NLRP3 [23–33]. How these different stimuli with various molecular patterns can be recognized by only NLRP3 is largely unknown. K+ efflux due to cell membrane disruption or lysosomal destabilization after crystal phagocytosis has been speculated to produce reactive oxygen species (ROS) that damage mitochondria, resulting in the activation of NLRP3 [34]. This activation leads to signaling through the adaptor protein ASC to activate pro-caspase-1, which then cleaves pro-IL-1β and pro-IL-18 into their active forms, resulting in inflammation (Fig. 16.2). NLRP3 activation is also known to cause necrotic cell death in hematopoietic cells and the subsequent release of danger signals, similar to the hyperresponse caused by R-proteins in plants [2, 3, 35]. NLRP3 is mainly expressed in hematopoietic cells such as LCs, DCs, and neutrophils in the skin, but also in other cell types, such as skin keratinocytes and mast cells [36, 37].
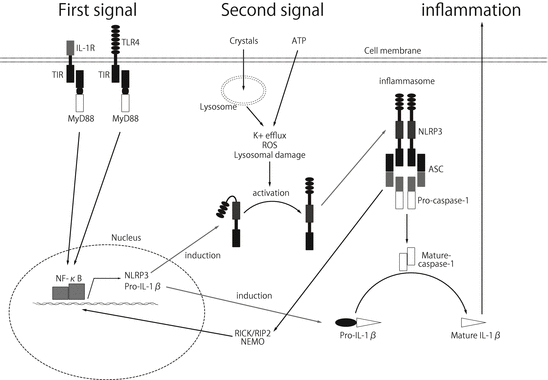
Fig. 16.2
The assembly of the NLRP3 inflammasome is known to require two distinct signals. (1) First signal: stimulation of PRRs, such as TLR4 and IL-1, activates the MyD88-dependent pathway via TIR. Activation of TIR results in NF-κB activation, leading to the upregulation of pro-IL-1β and, most importantly, the expression of NLRP3. (2) Second signal: NLRP3 can be activated by endogenous and exogenous stimuli, such as ATP and silica. The mechanism by which these ligands activate NLRP3 is largely unknown, but the involvement of potassium efflux, lysosomal damage, mitochondria damage, and reactive oxygen species (ROS) have been suggested. Upon activation of NLRP3, homotypic interactions are formed between the PYD domains of NLRP3 and ASC. Subsequently, the CARD domain of ASC molecules recruits pro-caspase–1, and active caspase-1 is produced, resulting in processing the pro-form of IL-1β into the active form of IL-1β. Additionally, the assembly of the NLRP3 inflammasome leads to the activation of RIP2, resulting in the activation of NF-κB and the production of pro-inflammatory cytokines through the ubiquitination of NEMO
Absent in melanoma 2 (AIM2) , a member of the PYHIN family (also called the HIN-200 family) and a cytosolic microbial dsDNA receptor, forms an inflammasome with ASC to trigger caspase-1 activation [38, 39]. AIM2 is expressed in LCs in normal epidermis and functionally expressed in keratinocytes under inflammatory conditions [40].
16.5 Other PRRs
RLRs contain three cytoplasmic RNA helicases that are critical for host antiviral responses [41]. RIG-I and MDA-5 sense dsRNA, leading to production of type I interferon. LGP2 contains an RNA binding domain, but lacks the CARD domains, and thus acts as a negative feedback regulator of RIG-I and MDA-5. In human keratinocytes, RIG-I is induced by IFN-γ and tumor necrosis factor (TNF)-α stimulation [42].
16.6 Antimicrobial Peptides
Pathogenic microorganisms in the skin can be killed by antimicrobial peptides (AMPs) , such as defensins and cathelicidins , which are constitutively produced by keratinocytes and induced after an inflammatory response, and direct activation of PRRs by microbial components [44]. They are short cationic peptides that disrupt the cell membranes of bacteria, fungi, and the membrane envelopes of some viruses within minutes. AMPs are an ancient, evolutionarily conserved class of antimicrobial peptides made by many eukaryotic organisms, including mammals, insects, and plants. AMPs have pleiotropic functions, not only to kill microbes but also to control host physiological functions such as inflammation, angiogenesis, and wound healing.
The two major subfamilies of AMPs in humans are cathelicidins and human β-defensins (hBDs) [45] (Table 16.2). The expression levels of hBD-2, hBD-3, and human cathelicidin in keratinocytes are very low at the steady state and upregulated during infection, inflammation, and wound healing [46, 47], whereas hBD-1 is constitutively expressed in keratinocytes [48]. Most AMPs have an overall net positive charge that ensures their accumulation and subsequent penetration of the negatively charged microbial cell walls [49]. In addition to keratinocytes, eccrine cells, sebocytes, neutrophils, eosinophils, mast cells, platelets, and T cells produce AMPs [50].
Table 16.2
Antimicrobial peptides (AMPs) and their sources
AMP | Examples | Structure | Cells |
|---|---|---|---|
α-defensin | α-defensin 1-4, HD-5,6 | β-sheet | Neutrophils, epithelial cells (GI) |
β-defensin | hBD 1-4 | β-sheet | Neutrophils, epithelial cells (keratinocytes, respiratory, GI) |
Chathelicidin | hCAP-18/LL-37 | α-helix | Neutrophils, epithelial cells (keratinocytes, respiratory, GI), mast cells |
16.7 Innate Immunity and Skin Diseases
Atopic dermatitis (AD): AD patients have a greater susceptibility to bacterial, fungal, and viral infections, such as S. aureus, C. albicans, and herpes simplex. Associations between genetic variants in TLR2 and severe AD [51–53] and reduced TLR2 expression in keratinocytes and circulating monocytes from patients with AD [54, 55] have been reported. TLR2 recognizes lipopeptides and lipopteichoic acid from S. aureus [56] and cell wall polysaccharide mannnan from C. albicans [57]. In addition, LL-37 (processed human cathelicidin hCAP-18) and hBD-2 are significantly decreased in skin lesions in AD patients [58], resulting in S. aureus colonization. Reduced LL-37 expression in AD also renders patients susceptible to eczema herpeticum caused by herpes simplex proliferation [59].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


