Innate
Adaptive
Early response to foreign antigens
Occurs within days of infection
Rapid response is the first line of defense
Antigen specific
Broad spectrum (not antigen specific)
Develops immune memory
No memory or long-lasting protective immunity
Aids recovery from viral infection as well as preventing re-infection
Key players include T and parenchymal cells and phagocytic dendritic cells
Key players include T and B lymphocytes
Some of these TLRs involved in the recognition of viruses have been identified. An example is TLR9, which recognizes HSV DNA on DCs and induces antiviral mechanisms that include the secretion of type I interferons (IFNs) [1, 6]. The interactions between the TLRs and the virus are necessary to guide, in this case, the anti-herpes immunity toward an adaptive (specific) cellular response (T-helper-1 [Th1] type, see below) [7]. Other examples of TLR interactions and viruses include the production of IFN-β and different chemokines activated by TLR3 and the induction of phagocytosis and inflammation by TLR4 mediated by the secretion of IFN-β [6].
The adaptive response is specific for antigen recognition, occurs within days of the infection, and is the result of the interaction of T and B lymphocytes. The immune players are multiple and capable of developing a lasting immune memory. This response not only aids recovery from the primary viral contact but also protects against re-infection. T lymphocytes recognize a processed antigen (short peptides) bound to the major histocompatibility complex (MHC) of the antigen-presenting cells (APCs). There are two major subsets of T cells: CD4 and CD8. CD4 cells recognize antigens bound to MHC class II (exogenous antigens taken from the extra- cellular milieu and processed in the endosome of the APC), while CD8 cells recognize the antigens bound to MHC class I (usually endogenous antigens) [3]. The activation of the CD4 cell results in the secretion of a variety of cytokines. Depending on the pattern of cytokine expression, the immune response is characterized as either a Th1 or Th2 response. Th1 cells secrete IFN-γ, which activates macrophages, natural killer cells, and cytotoxic CD8+ T lymphocytes (cell-mediated immunity). On the other hand, Th2 cells secrete mainly interleukin-4 (IL-4) and IL-10, helping the primed B lymphocytes to differentiate into plasma cells and secrete antibodies (humoral response) [3]. Also, an additional type of T cell has been shown to play an important role in the immune response against viruses. These cells, designated the regulatory T (Treg) cells, carry the CD4+ and CD25+ antigens. Treg cells recognize self antigens and prevent autoimmunity responses, regulate the responses to exogenous antigens, and are involved in the chronic and latent phases of viral infections [8, 9].
Viruses have developed a diversity of mechanisms to evade both the innate and adaptive immune response and thus establish an infection (or persist at least until a new progeny of viruses is released). Some of the cutaneous viruses share common evasion mechanisms and others have specialized systems to survive and become latent until they have a new opportunity to flare. Several of these mechanisms are discussed in detail in the sections that follow.
Human Herpes Virus
The human herpes simplex virus (HSV) types 1 and 2 are neurotropic viruses from the α-herpesvirus family. These viruses have a large molecular weight and harbor double-stranded DNA. The genome is in an icosahedral capsid, which is protected by a proteinaceous layer (tegument). The capsid is surrounded by a lipid bilayer with glycoproteins (envelope) [10]. Herpes simplex virus is distributed worldwide, affecting developed and developing societies. Animal vectors for human HSV have not been described, and humans appear to be the only reservoir [11]. Usually the first infection is asymptomatic, but this depends on the age and immune status of the host, the amount of the infective dose, and the presence of innate defenses that may abort the infection [12, 13]. About 80 % of the adult population in the developed world becomes seropositive to HSV-1 and more than 20 % are seropositive to HSV-2. People who experience a primary infection with one or more herpes viruses carry these viruses for the rest of their lives (usually in a latent state). The virus is retained in specific neural reservoirs and may become active with periodic episodes of viral replication and shedding [7].
Herpes simplex virus infection usually initiates in the mucosa. The virus replicates in epithelial cells and then enters the nervous system through the nerve termini. Control of the acute (and persistent) HSV infection involves the activity of natural killer (NK) cells, virus-specific CD4+ and CD8+ cells, IFNs, and virus-specific antibodies [14]. After entry into the mucosa and skin, HSV establishes a lifelong persistence in the neurons of the sensory ganglia. Herpes simplex virus persistence and latency have been demonstrated in human trigeminal, facial, and vestibular ganglia, and reactivations from these locations can cause herpes labialis, vestibular neuritis, and cranial nerve disorders among others [15]. The mechanisms of viral reactivation are not fully understood but are associated with different events such as ultraviolet (UV) light, stress, fever, infections, and immunosuppression [16]. Recent studies have demonstrated that certain neurons are more prone to productive infections versus other neurons that are more likely to exhibit viral latency indefinitely. This susceptibility appears to be governed by regulatory RNAs and other regulatory proteins yet to be identified [17]. Furthermore, it has been postulated that UV light triggers reactivation by inducing apoptotic signals and uncoordinated lytic gene expression [18]. The role of the immune system during the latent phase is crucial to maintain the virus under control [19].
Immune Response
Once the virus is in contact with host cellular receptors, the processes leading to viral infection are triggered. Glycoproteins in the lipid bilayer allow the viral envelope to fuse with the epithelial plasma membrane. Viral proteins are released into the cytoplasm and viral DNA enters the nucleus. This process triggers three phases of the innate response: (1) secretion of immune proteins, such as complement and natural antibodies; (2) an early-induced response, in which the main mediators are IFNs produced by the infected epithelial cells and resident DCs; and (3) the activation of inflammatory cells, such as neutrophils, macrophages, and NK cells [20]
At the site of mucosal contact the virus usually encounters several barriers, including mucus, normal bacterial flora, the glycocalyx, complement proteins, and natural immunoglobulin (IgM) antibodies [1, 20]. Although these substances act in concert to decrease the number of infected cells, HSV usually replicates successfully and triggers the early innate immune response. Humans may express different levels of natural antibodies to HSV, which is a reflection of their own past exposure experience [7, 20]
The early innate immune response has two goals: (1) limit viral replication and spread of the virus in uninfected cells, and (2) recruit other inflammatory cells [20]. The viral interaction with the epithelial cells may stimulate cell-surface TLR2 (e.g., in the genital mucosa) [1, 20]. With the activation of this receptor, the epithelial cells activate complement, chemokines, and IFN-α and -β soon (between 8 and 12 h) after the infection. Interferon-α and -β are produced by most cells types, but the DCs are responsible for the majority of their production. Interferon-α and -β are known to be two of the most important molecules to control HSV infection at this stage. Some studies suggest that resistance or susceptibility to HSV infection is directly correlated with the amount of IFN-α and -β produced [12].
Complement, chemokines, and IFN-α and -β activate the endothelial cells that express IL-8, tumor necrosis factor-α (TNF–α), IFN-γ, and granulocyte-macrophage colony-stimulating factor (GM-CSF), leading to neutrophil chemotaxis. Figure 17.1 illustrates the chemokine regulation of leukocyte movement [
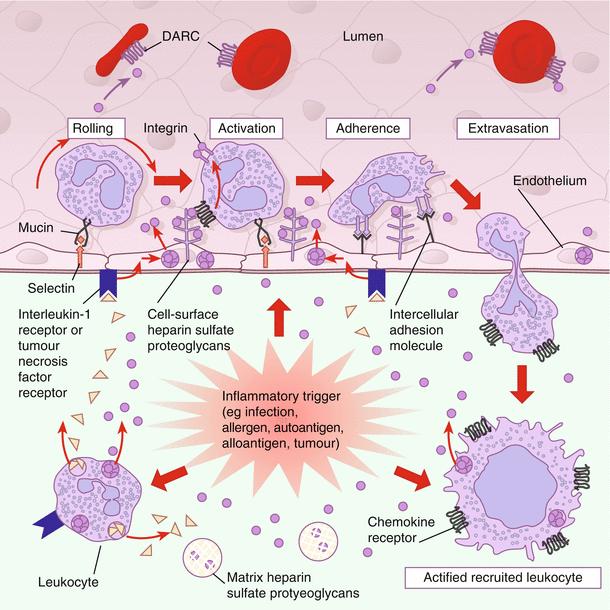
21]. The inflammatory reaction alerts the DCs and resident macrophages to the presence of the virus and induces a “state of awareness” or “antiviral status” in the uninfected cells [7]. Infected macrophages that are able to survive acute HSV infection become a significant source of inflammatory chemokines and cytokines including TNF-α, IL-1, IL-6, IL-8, IL-12, IL-18, RANTES (regulated on activation, normal T-cell expressed and secreted; a chemokine that is a chemoattractant for eosinophils, monocytes, and lymphocytes), as well as IFN-α and -β. Subsequently, the immature DCs capture viral antigens and transport them to the regional lymph node to alert and stimulate the adaptive immune response [7].
Fig. 17.1
Chemokine regulation of leukocyte movement. Chemokines are secreted at sites of inflammation and infection by resident tissue cells, resident and recruited leukocytes, and cytokine-activated endothelial cells. Chemokines are locally retained on matrix and cell-surface heparin sulfate proteoglycans, establishing a chemokine concentration gradient surrounding the inflammatory stimulus, as well as on the surface of the overlying endothelium. Leukocytes rolling on the endothelium in a selectin-mediated process are brought into contact with chemokines retained on cell-surface heparin sulfate proteoglycans. Chemokine signaling activates leukocyte integrins, leading to firm adherence and extravasation. The recruited leukocytes are activated by local proinflammatory cytokines and may become desensitized to further chemokine signaling because of high local concentrations of chemokines. The Duffy antigen receptor for chemokines (DARC), a nonsignaling erythrocyte chemokine receptor, functions as a sink, removing chemokines from the circulation and thus helping to maintain a tissue–bloodstream chemokine gradient. (From Luster [21]. Copyright © 1998 Massachusetts Medical Society. All rights reserved.)
Once the DC exits the mucosa, the late-induced innate response begins at the infection site. Initially, an influx of neutrophils, monocytes, and NK cells is established. These cells traverse the activated capillary endothelial cells guided by the chemokines present at the infection site. The neutrophils secrete α-defensins that insert into the virion lipid envelope and trigger the degradation of phagocytosed virions [22]. They also secrete TNF-α, which acts synergistically with IFN-α and -β, as well as IFN-γ to produce lysis of the infected cells inhibiting viral replication [22]. Simultaneously, NK cells are activated by the binding of immunoglobulins to the viral antigen (through their Fc receptor [CD16] located on the cell surface) and are recruited by chemokines and cytokines (IFN-α and -β, IL-12, IL-15, IL-18) produced by the activated infected cells within 2–3 days after infection. The NKs participate in cytolysis of virus-infected cells by perforin/granzyme-mediated processes that result in phagocytosis and destruction of infected cells and viral particles [20]. The accumulation of viral antigens is followed by complement activation, facilitating the uptake of viral peptides by phagocytes [23]. These phagocytic cells produce antiviral cytokines and defensins such as nitric oxide (NO), which enhance the immune response and promote phagocytosis and destruction of virus particles and infected cells [20].
One of the key cells for the innate response against HSV is the macrophage. The molecular mechanisms of the immune response within macrophages have been extensively studied. Once HSV infects macrophages, these cells release a series of cell mediators such as TNF-α and IL-12 (which subsequently stimulates the production of IFN-α and -β). Interferon-α and -β induce the NK and T cells to secrete IFN-γ, which eventually controls the HSV replication and initiates the adaptive response [24, 25]. At this stage, the site of origin of IL-12 production is controversial. The majority of investigators suggest that DCs are the main producers of IL-12 [7, 26]. However, other authors indicate that the main source of this molecule is the recruited inflammatory cells [27].
The adaptive immune response begins when the DCs migrate to the regional lymph nodes. These cells mature and display viral peptides coupled with MHC molecules, secrete cytokines, and regulate the expression of other inflammatory molecules. The cytokines produce differentiation of the Th0 cells to either Th1 or Th2. For HSV, the Th1 response is indispensable for clearance of the infection (particularly for HSV-2) [28]. Other cells such as NK cells and macrophages also stimulate the switch from a Th0 to a Th1 response [28] (Table 17.2).
Table 17.2
Immune responses to herpes viruses
Complement-mediated activation of immune cells |
Antibody mediated activation of immune cells |
Secretion of interferons (IFNs) by the infected epithelial cells |
Activation of neutrophils, macrophages, and natural killer (NK) cells |
Activation of endothelial cells with secretion of tumor necrosis factor-α (TNF-α), interleukin-8 (IL-8), IFN, and granulocyte-macrophage colony-stimulating factor (GM-CSF) |
Secretion of α-defensins by neutrophils |
Activation of NK cells for cytolysis of virus-infected cells |
Secretion of IL-12 and TNF-α by monocytes |
Differentiation into Th1 and Th2 responses |
One of the most studied T cells during HSV infection is the CD8+ T cell. These cells are capable of expressing the lymphocyte-associated antigen (cutaneous leukocyte antigen [CLA]) [29, 30]. During recurrent and symptomatic infection, antigen-specific CD4+ T cells and NK cells infiltrate the dermis by day 2 of the lesion formation, whereas CD8+ dermal infiltration (which implies cytotoxic activity resulting in viral clearance) occurs a few days later [29, 30]. Therefore, CD8 cells appear to play an important role in containing HSV infection. Posaved et al. showed that the levels of CD8+ cytotoxic T lymphocytes (CTLs) correlate inversely with the severity of HSV-2 infection and temporally with the local clearance of the virus in lesions [31]. The data from the study of Koelle et al. suggest that the expression of the homing receptor (CLA) by the CD8+ T cells is programmed at the site of the original antigen encounter and promotes migration and immune responses in reactivation [29]. Additional studies indicate that this specific cell has the capacity of T-cell memory and self-renewal that is crucial to control the infection and symptomatic recurrences [31].
Studies in vivo (mouse model) have demonstrated that CD8+ cells also play a crucial role in controlling the virus during the latent state [19, 32]. Latent herpes viral infection in humans in the trigeminal ganglia is accompanied by a chronic inflammatory process that involves elevated levels of cytokine transcripts such as IFN-γ, TNF-α (which affects viral replication), and chemokines, accompanied by persistent lymphocytic cell infiltration (CD8+ T cells, CD68+, and macrophages). It has been suggested that this phenomenon could be due to a low level of expression of viral genes during latency. The CD8+ T cells are capable of controlling the virus through cytokines, and this mechanism is likely to occur in response to the persistence of viruses that are prone to reactivate frequently, providing a survival advantage for both the host and the virus [19]. It is proposed that factors compromising CD8+ T cell function lead to virus escape from immune regulation and reactivation [33].
Immune Evasion Mechanisms
There appears to be a complex and well-balanced interaction between host cells and HSV. Success for the virus means a delicate balance between infection and latency. Therefore, evasive strategies must be carefully designed to permit viral replication at certain stages that could potentially lead to transmission to other susceptible hosts in order to maintain the viral progeny. Humans and the herpes viruses have evolved together in a symbiotic relationship that has allowed the adaption of the virus to the host and the environment, maintaining a “truce” with the immune system without any serious threats to human life. Some of the identified mechanisms to evade the immune system include the ability of HSV to alter the activity of neutrophils, macrophages, or NK cells [31]. It is known that HSV interferes with monocyte function, suppresses chemotaxis and phagocytosis, and reduces the secretion of IL-1 and TNF-α [7, 20, 34]. It has also been reported that HSV reduced the activity of NK cells upon contact with infected cells, due in part to the interference with IFN-α and -β production [20, 34]. Furthermore, HSV produces many antiapoptotic factors, such as cathepsins and latency-associated transcripts (LATs), which allow for maximal viral replications. LATs have been recovered from latently infected neurons and have been shown to inhibit granzyme-B mediated apoptosis and capsase-8-dependent apoptosis [17]. It has been proposed that LATs increase the efficiency of latency and subsequent reactivation [33] (Table 17.3).
Table 17.3
Mechanisms of immune evasion in herpes viruses infection
Modulation of the activity of neutrophils, macrophages, or natural killer (NK) cells |
Interference with monocyte function |
Suppression of chemotaxis and phagocytosis |
Modulation of the production of IL-1, TNF-α, IFN-α and -β |
Reduced activity of NK cells |
Inhibition of apoptosis |
Vaccines
There are currently two different groups of vaccines targeting HSV: prophylactic and therapeutic. Prophylactic vaccines aim to protect seronegative patients against HSV infection. The goal of therapeutic vaccines is to reduce symptoms and infectivity (outbreaks and shedding) in patients who are already seropositive for HSV [35]. In the recent decades, the focus has been primarily on developing a prophylactic vaccine with varying results in trials. Two studies were carried out by GlaxoSmithKline, using a recombinant vaccine preparation containing HSV glycoproteins. The first enrolled patients who were seronegative for both HSV-1 and HSV-2. The second study allowed inclusion of HSV-1–seropositive patients [36]. The studies’ primary end point was protection against genital HSV-2 clinical disease and the secondary end point was HSV-2 seroconversion. The first study did not show any difference in the primary end point between the group that received vaccine and the group that received a placebo. However, when a gender-stratified analysis was performed, the vaccine proved protective in females only (73 %; 95 % confidence interval [CI], 19–91 %). The sex differences found in these studies might be explained by innate anatomic differences of the genital epithelial layers between men and women. The epithelial layer of male genitals consists mostly of stratum corneum, which is lacking in the genitals of women at the vaginal-cervical mucous membrane. Additionally, the secretions produced by the vagina and the genital mucous membranes contain antibodies and migratory leukocytes. Therefore, women appear to have enhanced immune responses mediated by helper T cells exhibiting a Th1 response, compared to men [37]. The Herpevac Trial for Woman was conducted to evaluate this sex difference in vaccine protection. Although the primary endpoint of HSV-1 and HSV-2 genital disease prevention was not achieved, the vaccine was found to be effective in decreasing infection caused by HSV-1 but not HSV-2 [38]. Further studies are needed to evaluate this unexpected outcome.
Recent HSV vaccine research has transitioned away from prophylactic vaccines to therapeutic vaccine candidates. There are currently three novel HSV vaccine candidates in phase I or II trials. The HerpV vaccine is composed of recombinant human heat shock protein-70, 32 different HSV-2 antigens and an adjuvant. This vaccine increased HSV-2 CD4+ and CD8+ T cell activity in HSV-2 seropositive patients in a phase I trial [39]. It is currently being evaluated for efficacy in prevention of shedding and lesions in a phase II trial [40]. The GEN-003 vaccine consists of the gD2 glycoprotein, ICP4 (a potent CD8+ T-cell immunogen) and Matrix M adjuvant. In animal models, this vaccine candidate decreased recurrence of lesions and viral shedding [41]. The GEN-003 vaccine has completed a phase I/II trial evaluating its efficacy as a therapeutic vaccine but the results of this trial have not been published [42]. Finally, the HSV529 vaccine is composed of a DNA replication defective HSV virus. This candidate vaccine has demonstrated efficacy in animal models and is in a phase I/II trial as a therapeutic and prophylactic vaccine [43]. Research on a prophylactic HSV vaccine needs to focus on inducing a stronger antibody response. In contrast, research on a therapeutic HSV vaccine needs to focus on prompting a more potent T-cell response [43].
Varicella-Zoster Virus
Varicella-zoster virus (VZV) is also an α-herpesvirus with a genome of ~125,000 base pairs (bp) with at least 70 unique open reading frames (ORFs) [44, 45]. It is characterized by a relatively short reproductive cycle, rapid cell-to-cell spread, and significant ability to establish latent infections (primarily in sensory ganglia) [45]. Humans are the only known natural reservoir for VZV. This virus causes two different clinical syndromes: varicella (chickenpox) and herpes zoster (shingles). Varicella is usually a self-limited disease of childhood characterized by a very pruritic rash. The disease has a worldwide distribution, and transmission is more pronounced in temperate climates (where it is seen more frequently in children) than in tropical environments [11]. Approximately 20 % of those who had varicella later develop herpes zoster, which usually affects adults older than 50 years of age, although it can occur at any age [11, 46].
Immune Response
Initial clinical observations indicate that the primary site of inoculation is the respiratory tract. A rash usually develops after an incubation period of 10–12 days [11, 45, 46]. The entry of the VZV into a host cell requires fusion of the viral envelope with the host cell plasma membrane. This phenomenon is mediated by interactions of viral oligosaccharides with the heparan sulfate proteoglycans (via N-linkage) on the host cell surface. The fusion permits the entry of the viral proteins into the host cell cytosol and then to the nucleus where the nucleocapsid fuses with the outer nuclear membrane, releasing the viral DNA genome into the nucleus [47]. The spread of the virus is controlled by the innate and adaptive responses. The host cell membrane has specific glycoproteins such as gH, gL, gB, and gE that permit the fusion process between cells. These proteins are used by VZV to quickly spread to adjacent epidermal cells by inducing the fusion of the infected cells with noninfected ones [32]. Varicella-zoster virus has a special tropism for three major cell types: the peripheral blood mononuclear cells (PBMCs), skin cells, and sensory neurons [32]. This virus not only infects these cells, but also is capable of replicating inside them. This phenomenon of viral replication can be observed in intraepidermal vesicular lesions of the skin, which are loaded with free virus [11, 45, 47].
The virus infects the immature DCs of the respiratory mucosa, and these cells transport the virus to the T-cell–draining lymph nodes. From these nodes VZV is subsequently transported to the reticuloendothelial system and then is capable of gaining access to the bloodstream, causing a primary viremia. During this first viremic phase, VZV is able to reach the reticuloendothelial organs where it undergoes a phase of viral amplification. It was previously thought that VZV reached the skin during a second viremic episode that occurred in the late incubation period [47]. However, studies of viral infection in mouse models now suggest that infected T cells from the tonsil area are capable of transferring VZV to the skin immediately after entering the circulation during the primary viremia and that the prolonged interval between exposure and the skin rash reflects the time required for the virus to become recognized by potent innate immune barriers such as IFN-α [44, 47].
Varicella-zoster virus preferentially infects the active memory CD4 T cells that express skin homing markers such as CLA and the chemokine (C-C motif) receptor 4 (CCR4). These T cells are usually programmed for immune surveillance and then may facilitate the transfer of VZV into the skin [44]. It appears that VZV does not trigger any early inflammatory response that might block the appearance of virus-filled vesicles at the skin surface [44, 47]. The initial viremia is necessary to ensure that enough cutaneous lesions are formed to transmit the virus efficiently to other individuals as a viral survival mechanism. The T cells trafficking through the skin activate VZV replication at this site, and this process permits infection of more T cells that will return to the circulation [44].
The innate immune response is induced during the acute VZV infection. This response involves the release of IFN-α and IFN-γ and the secretion of cytokines such as IL-6 (by monocytes) via TLR2 [48]. This cell-mediated immune response contains VZV replication and prevents the host from a systemic disease (including severe compromise of organs such as lungs, kidneys, and spleen). The adaptive immune response begins with the presence of CD4+ T cells and the release of IL-2, IL-10, IL-12, and IFN-γ by Th1 and Th2 cells [49]. These cytokines cause the proliferation of VZV specific CD4+ and CD8+ T cells, which express MHC class I and II molecules and recognize the viral glycoproteins gE, gH, and gI, with the subsequent killing of the VZV-infected cells [50]. During the presence of rash, many mononuclear infiltrating cells are present. Most of these cells express CD4 and CD8, including CD45RO+ memory cells, skin homing CLA, and CCR4+ T cells. During this period, the skin shows expression of E-selectin (a cell adhesion molecule and CD antigen that mediates neutrophil, monocyte, and memory T-cell adhesion to cytokine-activated endothelial cells), intercellular adhesion molecule 1 (ICAM-1, a cell-surface ligand involved in leukocyte adhesion and inflammation), and vascular cell adhesion molecule-1 (VCAM-1, a cytokine- induced cell adhesion molecule present on activated endothelial cells, tissue macrophages, and DCs), allowing the migration of the inflammatory cells. The humoral immune response has a reduced role in controlling the virus. Immunoglobulin G (IgG), IgM and IgA appear to respond to some viral proteins. It seems that these antibodies neutralize cell-free virions and contribute in the lysis of infected cells [44] (Table 17.4).
Table 17.4
Immune responses to varicella-zoster virus (VZV)
Acute VZV infection |
Release of IFN-α and IFN-γ |
Secretion of cytokines such as IL-6 (by monocytes) via Toll-like receptor 2 (TLR2) |
Release of IL-2, IL-10, IL-12, and IFN-γ by T-helper-1 (Th1) and Th2 cells |
Proliferation of VZV specific CD4+ and CD8+ T cells |
During skin rash |
Expression of E-selectin, intercellular adhesion molecule 1(ICAM-1) and vascular cell adhesion molecule (VCAM-1) |
Migration of inflammatory cells |
Reduced role of humoral response in controlling the virus |
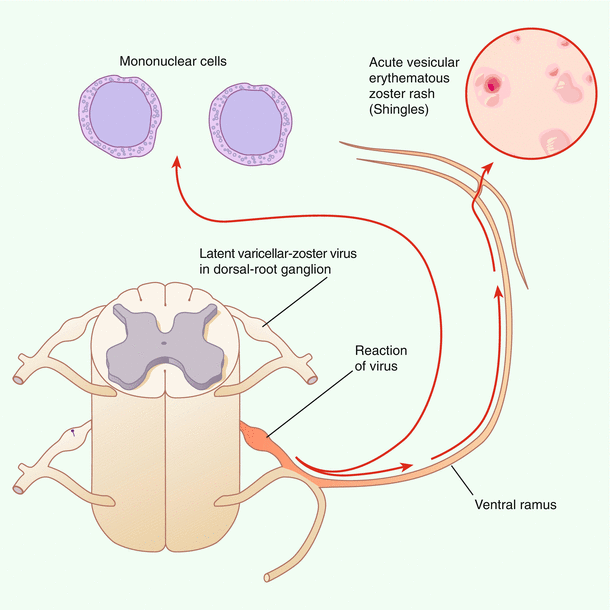
During the resolution of varicella, VZV establishes latency in the trigeminal and dorsal root ganglia where it remains latent through the lifetime of its host. Approximately 20 % of infected people develop herpes zoster. Figure 17.2 illustrates the latent virus in a dorsal root ganglion and reactivated virus causing acute vesicular zoster rash [
51]. In this period, the VZV multiplies and spreads centrifugally down the sensory nerve, causing neuronal necrosis and intense neuritis. Recent studies have shown that VZV possesses multiple open reading frames (ORFs) that encode proteins present in the cytoplasm of neurons during latency and are localized in the cell nucleus during reactivation [52]. Some of these genes are involved in VZV assembly, replication (such as ORF2) and latency. ORF47 encodes a protein that is part of the VZV virion tegument and is essential for VZV growth in differentiated skin and T cells [53]. Studies of this peptide indicate that the blockage of the kinase function of this protein decreases the VZV virulence in the skin, suggesting interference with the production of complete VZV virions. This peptide has also been implicated as a latency- associated protein along with the products of the ORF4 and ORF63 [54–57]. Additionally, the mechanisms of latency appear to be controlled LATs. However, the exact mechanisms are not well understood [58].
Fig. 17.2
Latent and reactivated varicella–zoster virus. Shown is a latent virus in a dorsal-root ganglion (white fusiform swelling) adjacent to the spinal cord and reactivated virus in a nearby dorsal-root ganglion (orange fusiform swelling) with transaxonal spread to the skin. The reactivated virus causes an acute, vesicular, erythematous zoster rash. (From Gilden et al [51]. Copyright © 2000 Massachusetts Medical Society. All rights reserved.)
Immune Evasion Mechanisms
Varicella-zoster virus utilizes several mechanisms to overcome the immune response. Major histocompatibility complex class II expression is restricted to APCs and is required to present the viral peptides to CD4 T cells. Usually, the host immune response to the virus produces the release of IFN-γ, which stimulates the expression of MHC II molecules. To evade the immune response and ensure replication, VZV produces a protein that interferes with Stat1 (a signal transducer and activator of transcription that mediates cellular responses to interferons). Stat1 interacts with P53 tumor suppressor protein and regulates expression of genes involved in growth control and apoptosis activation and thereby inhibits antiviral IFN-γ production in foci of infected skin cells [44]. Inhibition of IFN-γ results in decreased MHC II expression, which subsequently affects the efficiency of VZV specific immune responses. Varicella-zoster virus takes advantage of these circumstances, and as a consequence, the initial formation of VZV lesions in skin (varicella or during the VZV reactivation as zoster) is facilitated [44, 58, 59]. Additionally, VZV has mechanisms to delay clearance of virus-infected cells by interfering with the expression of MHC class I proteins that are necessary for CD4 and CD8 T-cell recognition [58]. This mechanism allows VZV to evade CD8 T-cell lysis during the viremic phase. The precise molecular event is related to the downregulation of MHC I by VZV via interference of its transport from the Golgi compartment to the plasma membrane [44] (Table 17.5)
Table 17.5
Immune evasion mechanisms in varicella-zoster virus infection
Interference with signal transduction mechanism through Stat 1 |
Decreased major histocompatibility complex class II (MHC II) expression |
Delay of clearance of virus infected cells through interference with MHC expression |
Vaccines
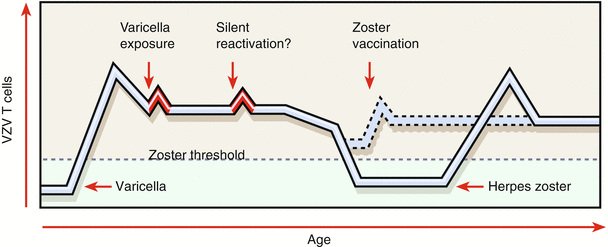
Before the introduction in 1995 of Varivax, a live attenuated Oka strain of VZV, millions of children developed primary varicella (chickenpox). Varivax is indicated for vaccination against varicella in individuals 12 months of age and older and it is recommended as part of the childhood immunization schedule. Its effectiveness is greater than 95 % for varicella prevention. Herpes zoster is the result of the reactivation of latent VZV infection residing in sensory ganglia. In the United States, herpes zoster affects approximately 1,000,000 persons annually, with an incidence of 2.2 cases per 1000 persons of age or older [60, 61]. Results from several clinical trials have determined that a live, attenuated VZV vaccine using the Oka strain (Zostavax) is safe, elevates VZV- specific cell-mediated immunity, and significantly reduces the incidence of herpes zoster and postherpetic neuralgia in immunocompetent people over the age of 50. This vaccine is 14-fold more concentrated than VARIVAX and has been approved for use in the United States. It is expected to reduce the risk for herpes zoster by >50 % [61]. There have been concerns about the decline in Zostavax efficacy however. Although vaccine efficacy was maintained through year 5 after vaccination in the Short-Term Persistence Substudy, it steadily declined during that time period [62]. Figure 17.3 illustrates how the administration of zoster vaccine to older persons may prevent VZV-specific T cells from dropping below the threshold for zoster occurrence [
63].
Fig. 17.3
Host factors in varicella-zoster virus (VZV) latency and reactivation. Varicella is the primary infection caused by VZV, and its resolution is associated with the induction of VZV-specific memory T cells (blue line). Memory immunity to VZV may be boosted periodically by exposure to varicella or silent reactivation from latency (red peaks). VZV-specific memory T cells decline with age. The decline below a threshold (dashed green line) correlates with an increased risk of zoster. The occurrence of zoster, in turn, is associated with an increase in VZV-specific T cells. The administration of zoster vaccine to older persons may prevent VZV-specific T cells from dropping below the threshold for zoster occurrence (dashed blue line). (From Arvin [63]. Copyright © 2005 Massachusetts Medical Society. All rights reserved.)
Human Papillomavirus
Human papillomaviruses (HPVs) are small DNA tumor viruses that have a circular double- stranded DNA genome of ~8 kb in length. They belong to a large group of recognized oncogenic viruses [64]. Human papillomavirus is a member of the Papillomaviridae family that includes over 120 different genotypes (defined by differences in their nucleotide sequence) [11, 65]. The HPV genome is composed of three regions: the long control region (LCR), the early region (E), and the late region (L). The early region consists of genes E1 to E8 (which encode nonstructural proteins for transcription, plasmid replication, and transformation), while the late region codes for the major (L1) and minor (L2) proteins that form the viral capsid. The current vaccines include the protein L1 as the major immunogenic antigen [66].
The HPVs are commonly categorized as having low or high oncogenic potential. Infections with the high-risk types such as HPV-16 and HPV-18 have been implicated in cervical-anogenital cancer and oral squamous cell carcinomas [11, 67, 68]. The infection of the genital tract by HPV is one of the most frequent sexually transmitted diseases [68, 69]. Approximately 75 % of sexually active individuals are infected by HPV during their lifetime. In the United States, it has been calculated that 6.2 million new cases of high-risk HPV infections occur each year, close to 20 million Americans are infected, and 1 % of sexually active adults have genital warts [65, 69]. It is estimated that >99 % of cervical, as well as >70 % of anal and vaginal cancers are related to HPV infection (cervical cancer is the second most frequent cause of death due to neoplasia among women worldwide). More specifically, about 30–40 % of penile, vulvar, and oropharynx cancers are related to the HPV-16 and HPV-18 [68, 70–72].
The majority of HPV clinical lesions are located in the genital area (70–90 %) in sexually active adolescents and young women. These lesions usually show clearance of the infection within 12–30 months [68]. Longer duration of infection is frequently related to the presence of cervical intraepithelial neoplasia (CIN) [73]. The prevalence and incidence of the HPV infection varies with age. The cause for this apparent variation is not clear. Many authors postulate that some (but not all) infected individuals develop an adaptive immune response against HPV that prevents future infections [74].
Immune Response
Human papillomavirus has a special tropism for the epithelial cells (they are epitheliotropic), infecting keratinocytes at a wide range of body sites where they cause aberrant cellular proliferation leading to benign warts or cervical cancer [3, 64, 75]. Initially, the virus infects primitive basal keratinocytes and starts DNA replication and transcription of the early genes at very low levels [76]. The peak of productive synthesis of virions is reached once the keratinocyte reaches higher strata of the epithelium. Thus, high levels of viral proteins and viral assembly occur only at the upper layers of the squamous epithelia (stratum spinosum and granulosum) [11]. At this point the cycle of replication and patterns of gene transcription are dependent on the stage of differentiation of the keratinocyte [76].
A significant amount of information regarding the immune response has been gathered from observations in animal models. Once the virus has reached the skin, it establishes itself at the basal cells of the epithelia where it starts replication. At this point the replication is minimal but becomes greater once the keratinocyte matures toward desquamation [77]. At the basal layer, there is practically no immune response against the virus (two to several months), but once it is detected by the immune system, replication increases and results in clinically detectable lesions [78, 79]. After a few months (which varies depending on the host), the infected keratinocytes express the late viral genes (encoding proteins L1 and L2) that trigger a full immune response [77–79].
The keratinocytes are able to secrete cytokines, growth factors, and chemokines upon viral stimuli. The host immune response dictates the emergence and characteristics of clinical lesions [76]. Some of the cytokines involved are transforming growth factor-β (TGF-β), TNF-α, and IFNs. TGF-β has been shown to inhibit the growth of nontumorigenic HPV-16 and HPV-18 immortalized cells, and it seems to control the expression of E6 and E7 (although some controversy exists on these facts) [76]. In normal cervical cells, TGF-β inhibits viral growth, but HPV can evade this control mechanism [76]. TNF-α, which is another product of the keratinocyte, may have an antiproliferative effect on the HPV-16–infected cells through cell cycle arrest (between the G0 and G1 phases of the cell cycle) [76, 80]. It has also been suggested that TNF-α acts as a repressor on the expression of E6 and E7 in the immortalized human keratinocytes [81]. The effects attributed to IFNs may be virus-type specific. These molecules also appear to inhibit the production of viral proteins [76].
The time between HPV infection and the development of a clinical lesion could vary from weeks to more than 9 months. This is an indication that HPV could modulate the immune system as evidenced by the following: (1) HPV infects mainly keratinocytes, which are cells programmed to die and desquamate in a specific and timely manner (apoptosis); and (2) the intracellular location of HPV in these cells allows the virus to hide and avoid immune surveillance. As a consequence, the apoptotic keratinocyte infected with HPV does not trigger a significant inflammatory reaction, eventually resulting in a persistent chronic infection [78, 82].
The adaptive immune response involves two phases: the recognition of antigen and the response to it. This adaptive immune response involves the LCs that capture the virus and its antigen for transport to local lymph nodes and presentation to naive T cells. The T cells return to the infected epithelial tissues via mechanisms that include secretion of chemokines and expression of adhesion molecules to clear the infection. In the recognition phase, the LCs are the major APCs. However, in some HPV infections, a depletion of these cells has been documented, which is associated with enhanced survival of HPV, prolonged courses of infection, and possibly malignancy [83–85]. At this stage, the infected keratinocytes produce IL-1α and TNF, which promote LC migration.
If the immune response is appropriate, the regression of warts usually follows. At this stage, a large infiltrate of T cells (CD4+ and CD8+) and macrophages is present, and the cytokines involved exhibit the Th1 pattern (IL-12, TNF-α, and IFN-γ plus the expression of the adhesion molecules for lymphocyte trafficking) (Table 17.6). A systemic T-cell response to E1 and E6 viral proteins is effective only at the peak of wart regression (although activity is present during the whole infective cycle) [78, 79].
Table 17.6
Immune responses to human papilloma virus
Secretion by keratinocytes of cytokines, growth factors, and chemokines (TNF-α, transforming growth factor-β [TGF-β], and IFNs) upon viral stimuli |
Langerhans cells capture of virus and its antigen |
Transport to local lymph nodes and presentation to naive T cells |
Large infiltration of T cells (CD4+ and CD8+) and macrophages |
Th1 pattern of cytokines (IL-12, TNF-α, and IFN-γ) |
Expression of the adhesion molecules for lymphocyte trafficking |
In summary, HPV is capable of escaping the immune response for several months by modulating the inflammatory reaction from the keratinocytes after invasion. Nonetheless, in the majority of cases, the immune system finally catches up and is capable of detecting the virus, (probably when the replication turnover is highest) and thus is able to resolve the infection.
Viral Oncogenesis
It is now accepted that HPV infection is necessary (but not sufficient) to cause cervical cancer, other anogenital neoplasms, and oral squamous cell carcinomas. Several viral types (such as 16 and 18) have been clearly identified as having high oncogenic potential. Low risk types include 6 and 11 [86]. Recently, significant evidence is emerging supporting the transition zone of the cervix as the location of persistent HPV infection. Specifically, it has been demonstrated that HPV exists in the latent state in the basal epithelial stem cells even after immune clearance of papillomas [87]. A recent animal model has shown that basal epithelial cells expressing the HPV 16 genes can undergo malignant transformation [88]. For high-risk HPV types, the proteins E6 and E7 appear to play a very important role in oncogenesis since they are able to inhibit two well-known oncogenic suppressor genes encoding the pRb (product of retinoblastoma tumor suppressor gene) and p53 proteins. E6 enhances the degradation of p53 via the ubiquitin-mediated proteolysis machinery (E3 ubiquitin ligase, UBE3A) and E7 interacts with the pRb [89]. Of note, Hyland et al. and McLaughlin-Drubin et al. recently demonstrated that E6 and E7 induce epigenetic reprogramming, specifically methylation/acetylation, modification of chromatin structure and subsequent activation of homeobox genes, in infected keratinocytes [90, 91].
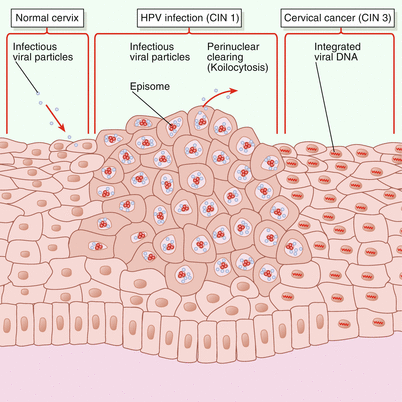
Many studies have shown that the absence of mature APCs, CD50, and CD86 and the downregulation of TNF-α represent an inadequate immune response to HPV that leads to persistence of the viral infection in the CIN lesions. The LCs are also decreased in numbers and lack the expression of CD11a/18, CD50, CD54, CD58, and CD86. These changes alter the antigen-presenting capacity and can induce immune tolerance to the viral infection [92]. The immortalization of epithelial cells in CIN produced by HPV-16 is related to the lack of response to the inhibitory effect of TGF-β and the inhibition in keratinocytes of TNF-α by the HPV-16 E7 protein, resulting in uncontrolled growth of the infected cells and escaping of the virus from the immune system [76]. Figure 17.4 illustrates the changes in cervical squamous epithelium caused by HPV infection [
93].
Fig. 17.4
Spectrum of changes in cervical squamous epithelium caused by human papillomavirus (HPV) infection. The left side of the figure shows normal cervical squamous epithelium. When HPV infection occurs (center), the virus exists in the cell nucleus as a circular episome. If the viral genome is intact, new infectious viral particles can be produced; their presence in the cell is indicated by perinuclear clearing, or koilocytosis. In cervical cancer (right), oncogenic portions of HPV DNA become integrated into the host’s DNA, with disruption of the E2 regulatory region and loss of other genes needed to form a complete virus. The cells are undifferentiated and do not show koilocytosis. CIN 1, cervical intraepithelial neoplasia grade 1. (From Goodman and Wilbur [93]. Copyright © 2003 Massachusetts Medical Society. All rights reserved.)
Immune Evasion Mechanisms
The reasons for the failure of the immune system to recognize HPV are not well understood. It appears that HPV is able to escape detection by the APCs. To survive and have enough time to replicate without detection, HPV has developed many strategies to evade the immune system. The special tropism for keratinocytes (as discussed earlier) is one of the first mechanisms of immune evasion [78].
Keratinocytes are powerful immune cells. They constitute almost 95 % of the cervical and skin epithelium and can express MCH II and co-stimulatory signals to T cells (such as ICAM-I) during inflammation [92]. If the immune response is activated, the HPV-infected keratinocytes release TNF-α, which negatively affects the replication of HPV [70, 92]. TNF-α upregulates the expression of ICAM-1 levels, which decrease the levels of IFN-γ. In CIN, the keratinocytes express less TNF-α, decreasing the stimulus to the LCs. This alteration also affects the expression of MHC II, and as a consequence the presentation of antigens is altered and the expression of the suppressive cytokine IL-10 is increased [68, 92]. These changes in the immunologic microenvironment (with inappropriate T-cell presentation) may contribute to the persistence and progression of the viral infection and development of a CIN lesion [84, 92, 94].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


