, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
19.1 Halogenodermas
19.1.1 Clinical Features
Halogenoderma is a rare dermatosis that results from the accumulation of halogenides following therapeutic exposure to bromide, fluoride, iodide, or iodine-containing products [1]. Occurrence appears to be greater in individuals unable to clear these compounds, such as those with acute or chronic renal failure [2]. Cutaneous lesions vary and may include acneiform eruptions, vegetative plaques, bullae, and ulcers [1–4]. With elimination of iodides and bromides, lesions slowly resolve over weeks, although resolution may be hastened by supplemental use of diuretics and corticosteroids [2, 5].
19.1.2 Histology
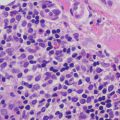
This group of disorders includes iododerma and bromoderma. The histologic features are quite similar, and they will be described together. The epidermis is hyperkeratotic and demonstrates pseudoepitheliomatous hyperplasia in some cases, or it can be entirely normal in others [6]. Within the dermis and occasionally within the epidermis, there is a brisk lymphohistiocytic infiltrate with abundant eosinophils and neutrophils (Figs. 19.1 and 19.2). Intraepidermal abscesses are frequently encountered [2, 4].
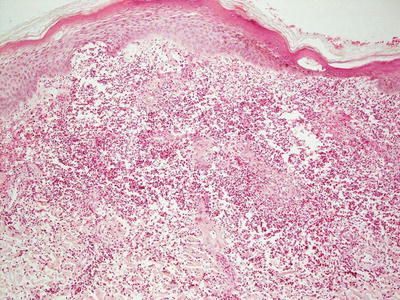
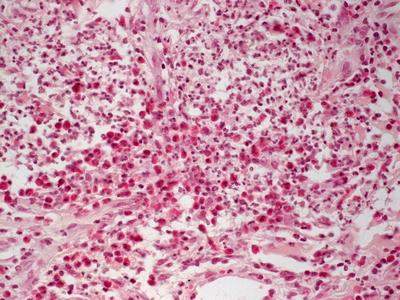
Fig. 19.1
A dense dermal infiltrate of neutrophils and eosinophils is present in iododerma
Fig. 19.2
Iododerma demonstrates abundant and diffuse dermal neutrophils and eosinophils
Halogen-induced panniculitis has been attributed to treatment with potassium bromide. A lymphocyte-mediated panniculitis with overlying ulceration was described. Unlike the majority of eruptions in halogenodermas, abundance of neutrophils and eosinophils was not noted in bromoderma. There have been no subsequent reports of a similar process, so if this occurs, it appears to be extremely uncommon [7].
The histologic differential diagnosis of halogenoderma is primarily based upon the epidermal hyperplasia. It includes deep fungal infection, mainly chromoblastomycosis, blastomycosis, and sporotrichosis. Special stains for microorganisms are helpful in excluding these infectious entities, along with the lack of significant granulomatous reaction in halogenodermas. Pemphigus vegetans is also in the differential diagnosis, but the acantholysis seen in that entity is not present in halogenodermas. Blastomycosis-like pyoderma also enters into the differential diagnosis. Observation of microorganisms or positive tissue cultures is helpful in resolving this dilemma. Squamous cell carcinoma and keratoacanthoma may demonstrate similar histologic changes, but cytologic atypia is more pronounced in these cases. Moreover, these tumors are uncommon in the pediatric population .
19.1.3 Pathogenesis
The pathogenesis of halogenoderma is unknown. Halogenoderma usually occurs in association with iodide and bromide ingestion as well as fluoride and chloride ingestion. Halogens such as lithium act at the level of the thyroid gland to block the release of thyroxine. There are reports of association between lithium use, and the development of goiter and hypothyroidism [8]. Lithium also inhibits iodide uptake in the thyroid gland [9].
19.2 Anticonvulsant-Induced Lymphoid Hyperplasia/Pseudolymphoma Syndrome
19.2.1 Clinical Features
Pseudolymphoma syndrome is a rarely reported cutaneous hypersensitivity to anticonvulsant therapy. Two to three weeks following initiation of the causative medication, patients develop skin lesions characterized by erythematous, firm subcutaneous nodules or papulonodules with the potential for progression to erythroderma [10]. Some reports highlight the minimal degree of concomitant systemic symptoms, while others describe associated fever, lymphadenopathy, arthralgia, transaminitis with hepatosplenomegaly, and blood cell dyscrasia, such as eosinophilia and, less commonly, leukopenia and agranulocytosis [10, 11]. Additionally described clinical features include a generalized morbilliform eruption, although this may be more appropriately qualified as DRESS syndrome (drug rash with eosinophilia and systemic symptoms), especially when striking systemic insult is noted [11]. Significant improvement is seen within weeks of cessation of the responsible anticonvulsant drug [10, 11].
19.2.2 Histology
Anticonvulsant therapy may result in a florid cutaneous lymphocytic hyperplasia [12]. While originally described secondary to phenytoin, a similar process also may occur secondary to treatment with other anticonvulsants [13, 14]. In some cases, the changes are strikingly similar to those seen in mycosis fungoides [13, 15]. There is slight epidermal hyperplasia with marked exocytosis of lymphocytes into the epidermis (Fig. 19.3). Spongiosis is present to varying degrees or may be virtually absent. Pautrier’s microabscesses may be present as well [11]. The lymphocytes are present within the superficial dermis, and may or may not extend deeper into the reticular dermis (Fig. 19.4). Scattered eosinophils are present in some but not all cases. In other cases, they may be abundant. Lymphocytes may appear atypical and enlarged.
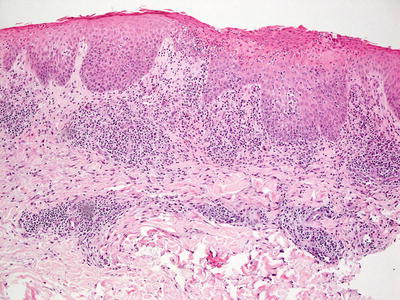
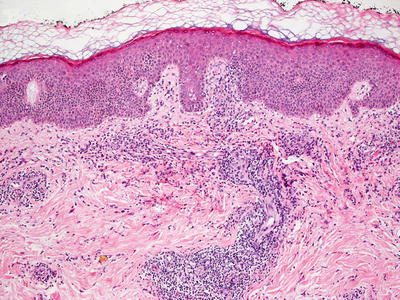
Fig. 19.3
Dilantin hypersensitivity reaction demonstrates a brisk lymphoid infiltrate, often with abundant exocytosis into the overlying epidermis in anticonvulsant-induced lymphoid hyperplasia
Fig. 19.4
A dense lymphoid infiltrate within the dermis and focally extending into the epidermis is seen in Dilantin hypersensitivity reaction in anticonvulsant-induced lymphoid hyperplasia
Immunophenotyping and genotyping are helpful in making the distinction from mycosis fungoides. In drug-induced eruption, there is a mixture of CD4+ and CD8+ T lymphocytes, while in mycosis fungoides, a marked predominance of one population (usually CD4+ cells) is seen [16]. Increased numbers of CD30+ cells are present in some cases of this drug-mediated process [14]. Clonal rearrangement studies are detected in most cases of mycosis fungoides, and would be unusual in drug eruption .
19.2.3 Pathogenesis
The mechanism of anticonvulsant-induced lymphoid hyperplasia is not known. It has been suggested that anticonvulsants, such as phenytoin, suppresses both cellular and humoral immune responses, and the loss of suppressor T-cell function may result in increased proliferation of clones of aberrant T lymphocytes [17–19].
19.3 Drug-Induced Hypersensitivity Syndrome and Protease Inhibitor-Induced Eruptions
19.3.1 Clinical Features
Drug-induced hypersensitivity syndrome (DIHS), also known as Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) , is a multi-organ reaction to a number of different medications. DRESS is classically defined by the onset of fever, rash, lymphadenopathy, and hepatitis. Symptoms typically begin 2–6 weeks following initiation of the inciting drug [20].
According to the European Registry of Severe Cutaneous Adverse Reaction study group, the bulk of cases (35 %) are secondary to aromatic antiepileptic medications, specifically carbamazepine, phenobarbital, and phenytoin, although there are reports of DRESS with valproic acid and zonisamide [21]. Antimicrobial sulfonamides and dapsone account for 12 % of cases, while other antibiotics account for 11 % of cases [21]. Individuals with certain HLA-haplotypes may be at increased risk for the disorder secondary to predisposition to impaired drug metabolism [20]. However, no consistent racial or gender predilection has been demonstrated [22].
Dermatitis is present in >90 % of affected individuals, beginning as an erythematous, morbilliform eruption of coalescing macules and papules at the face, upper trunk, and proximal extremities with distal spread and potential for erythroderma (Figs. 19.5 and 19.6) [22]. Pustules may be noted in darker-skin individuals. Pronounced facial edema and symmetric lymphadenopathy are key features (Fig. 19.7). Mucosal surfaces can be involved as well with conjunctivitis, “strawberry tongue,” pharyngitis, and oral ulcerations. However, the frank necrosis of erythema multiforme and Stevens Johnson syndrome/Toxic Epidermal Necrolysis are not frequently seen [22]. Hematologic abnormalities include eosinophilia, anemia as well as lymphocyte and platelet dysregulation [22].
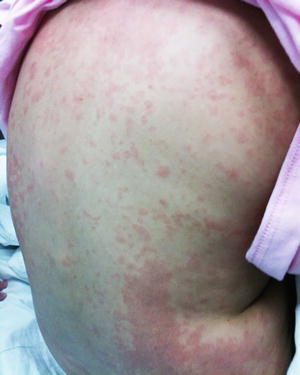
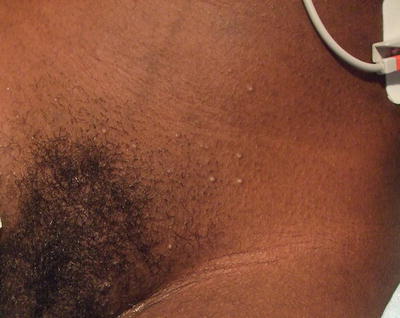
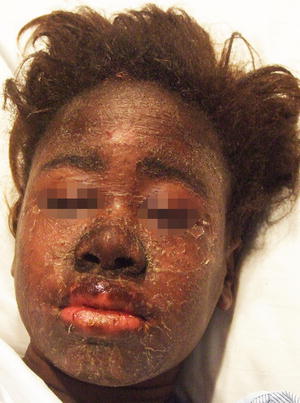
Fig. 19.5
DRESS presents as morbilliform eruption of erythematous papules and macules that coalesce along the trunk in a teenage patient who presented with lymphadenopathy, transaminitis, and eosinophilia weeks after starting a new antiepileptic medication
Fig. 19.6
DRESS may present in darker-skin with pustules interspersed within the morbilliform eruption. Erythema may be more difficult to appreciate
Fig. 19.7
Facial edema and cervical lymphadenopathy are key features of DRESS
Rapid recognition of the clinical features of DRESS and immediate discontinuation of the offending agent are key to clinical management in order to minimize symptoms and prevent more damaging sequelae. Further treatment is dependent upon the clinical progression, and the degree of systemic involvement. Skin changes may persist for weeks to months after stopping the offending drug with subsequent desquamation and possible post-inflammatory dyschromia .
Morbidity and mortality in DRESS are primarily due to hepatic disease, which was noted in 60–80 % of pediatric cases in one report [22]. Severity ranges from mild transaminitis and hepatomegaly to fulminant hepatic failure [22]. Renal manifestations (nephritis, hematuria, and proteinuria) were noted in 15 % of patients [22]. Acute and chronic thyroid dysregulation may be seen as well, warranting screening and monitoring even after resolution of symptoms. Pancreatitis, pneumonitis, pulmonary edema, myositis, and myocarditis have been reported in DRESS with only rare incidence of associated central nervous system dysregulation [22]. The mortality rate has been reported to be as high as 10 %, although death is less likely in children [21, 23].
19.3.2 Histology
Cutaneous eruption in DRESS syndrome demonstrates a superficial and deep perivascular inflammatory infiltrate with abundant eosinophils [27–29]. Epidermal spongiosis is a common finding, and scattered dying epidermal keratinocytes are present in a minority of cases [30] (Figs. 19.8 and 19.9). In some cases, eosinophils are not abundant in the skin lesions despite systemic eosinophilia [31]. This diagnosis requires clinical correlation, since it has no specific diagnostic histologic features.Protease inhibitors are responsible for inducing cutaneous eruptions with myriad histologic patterns. Nonspecific superficial and deep perivascular lymphohistiocytic infiltrate is a common finding [24, 25]. An interstitial dermal infiltrate is described in some patients, as well as an interface dermatitis with scattered dying keratinocytes and a superficial perivascular lymphocytic infiltrate [25]. Other presentations include neutrophil- and lymphocyte-mediated vasculitis [24, 26].
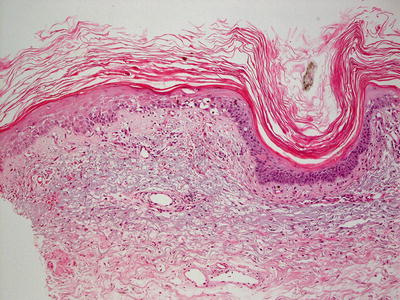
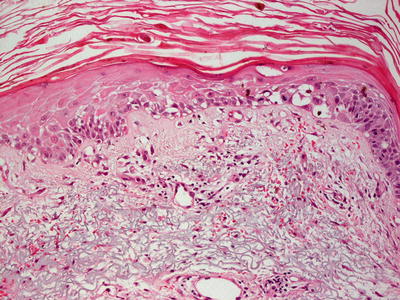
Fig. 19.8
An interface dermatitis with focal exocytosis of lymphocytes into the epidermis in a reaction to protease inhibitors in DRESS
Fig. 19.9
Extensive basal vacuolization is seen with a drug-induced reaction in DRESS
19.3.3 Pathogenesis
The pathogenesis of DRESS remains to be determined. However, DRESS is believed to be associated with the reactivation of the human herpesvirus family, particularly Epstein–Barr virus (EBV) and human herpesvirus-6 (HHV-6) [35–38].
Immunological reactions to certain drugs and HHV-6 reactivation appear to play a key role in the severity of DRESS. It has been suggested that detection of HHV-6 reactivation is a useful marker in the diagnosis and prognosis of this syndrome [38]. In the case of HHV-6 reactivation, there are increased HHV-6 antibody titers and virus DNA levels [37, 38]. Moreover, the levels of tumor necrosis factor-α (TNF-α) are elevated in early disease stage. It has been proposed that TNF-α levels may be a useful marker of the disease process because it is an early marker of HHV-6 reactivation [39]. Another marker that could be useful in distinguishing DRESS with HHV-6 reactivation from other drug reactions is serum TARC (thymus and activation-regulated chemokine)/CCL17 levels, which is elevated in patients with HHV-6 reactivation and not in those without [40].
Induction of CD8+ T lymphocytes in response to EBV infection, and the production of TNF-α and interferon-γ (IFN-γ) by activated T lymphocytes may contribute to the immunological response in DRESS [41]. CD8+ T lymphocytes are induced in the early stage of disease, followed by CD4+ T lymphocytes and FoxP3+ regulatory T (Treg) cells in the later stage [42]. The levels of FoxP3+ Treg cells are significantly increased in the skin lesions of DRESS as compared with other skin eruptions, such as graft-versus-host disease [43]. Moreover, the ratio of FoxP3+ Treg cells to CD3+ T lymphocytes is significantly higher in DRESS lesions. The increased number of FoxP3+ T cells that infiltrate skin lesions in DRESS may play a protective role in limiting the extent of epidermal damage in this disorder . Skin eruptions associated with protease inhibitors may be related to the interferon pathway. For example, the protease inhibitors telaprevir and boceprevir for the treatment of hepatitis C are targeted to NS3/NS4 proteases [32]. NS3/NS4 complex suppresses the activation of interferon regulatory factor-3 (IRF-3), which is a major transcription factor [33, 34]. IRF-3 promotes the production of interferon, and regulates many of the interferon-stimulated genes in response to viral infection. Protease inhibitors may increase interferon efficacy, but they also increase skin rash associated with interferon [32].
19.4 Radiation Dermatitis
19.4.1 Clinical Features
Skin reactions are a common and expected effects of radiotherapy. Acutely, dermatitis to radiotherapy is characterized by in-field erythema that evolves to desquamation that can last for weeks [44]. Localized erosions and hemorrhagic crusting can occur as well. Patients may complain of mild discomfort (tenderness or pruritus) at the affected area. Skin changes are graded from a level of 1 to 4 dependent upon the degree of erythema, edema, desquamation, and presence of necrosis, with grade 4 disease characterized by hemorrhagic ulcerations [45]. In patients with long-term unprotected sun exposure at treated areas, chronic changes may be seen months to years following therapy, most often characterized by some degree of poikiloderma [44]. The likelihood of secondary skin cancer is greater in those who have received ionizing radiation, with Caucasians and those with exposure at younger ages at greatest risk for cutaneous malignancy [45].
19.4.2 Histology
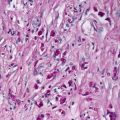
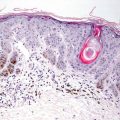
Early radiation dermatitis is characterized by the presence of dying keratinocytes, spongiosis, vascular thrombosis, and extravasation of erythrocytes [46]. In severe acute radiation dermatitis, a dense neutrophilic infiltrate and necrosis may be present. Neutrophils extend into the epidermis with concomitant apoptosis. Well-developed lesions demonstrate more characteristic changes. Hyperkeratosis overlies an epidermis that may be slightly atrophic. Spongiosis and basal vacuolization are present in some cases, but may not be apparent. Within the dermis, there is dense dermal fibrosis with elastotic degeneration of collagen surrounding dilated blood vessels (Figs. 19.10 and 19.11). Endothelial cells are often edematous. A perivascular lymphocytic infiltrate and fibrin thrombi within blood vessels results in a pseudovasculitis appearance [47]. A useful diagnostic clue is the presence of radiation fibroblasts, which are enlarged and hyperchromatic fibroblasts seen coursing between collagen bundles throughout the dermis. A pattern of subacute changes that resembles those present in cutaneous graft-versus-host disease has been described. In these cases, an interface dermatitis with abundant dying keratinocytes and lymphocytes is present [46].
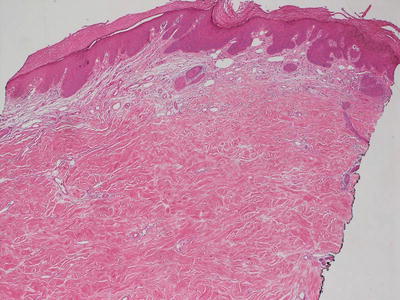
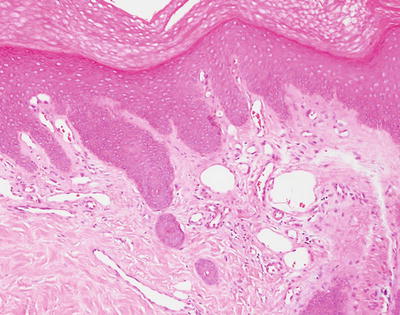
Fig. 19.10
Dense dermal sclerosis characterizes the changes seen in radiation dermatitis
Fig. 19.11
Vascular ectasia and some endothelial cell prominence are seen in radiation dermatitis
19.4.3 Pathogenesis
Ionizing radiation induces dermal and vascular injuries [45]. Transforming growth factor (TGF)-β plays an important role in the development of chronic radiation dermatitis. Ionizing radiation activates TGF-β, which in turn promotes the differentiation of skin fibroblasts into myofibroblasts. TGF-β induces the production of extracellular matrix proteins (such as collagen, fibronectin and proteoglycan) and inhibits the degradation of these proteins, leading to tissue fibrosis [48–51]. Studies of individuals exposed to radiation showed increased collagen production in cells from skin after a single dose of radiation in comparison with nonirradiated skin [52]. In animal models with defective TGF-β pathway, the animals have less tissue fibrosis after irradiation, and have increased tissue-healing processes [53]. Conversely, upregulation of TGF-β has been observed in fibrotic tissue of irradiated patients but not in nonirradiated controls [49]. These findings confirm the role of TGF-β in cellular and tissue responses to radiation. In addition, endothelial cell and vascular damages due to radiation can activate the coagulation system, which in turn promotes cytokine production and leads to a robust inflammatory response and matrix deposition [45, 54]. Importantly, radiation-induced tissue injury can result in cutaneous malignancies later on, such as basal cell carcinoma, squamous cell carcinoma, fibrosarcoma, and angiosarcoma , but these complications are rarely seen during childhood [45, 55].
19.5 Striae Distensae
19.5.1 Clinical Features
Striae distensae are a common finding in the general population, most notably in adolescents. They occur in approximately 35 % of girls and 15 % of boys between 9 and 16 years old [56]. Predisposing factors include rapid growth, obesity, puberty, pregnancy, positive family history, Cushing syndrome, and prolonged use of corticosteroids (both systemic and potent topicals) [57].
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


