Fig. 1.1
Medication research involves a “Life Cycle” which starts with observation of a clinical or basic science phenomenon. The observation leads to testable hypotheses, which point the way to a biological mechanism of disease. Confirmed hypotheses lead to potential diagnostic tools or therapies which are further tested and refined in preclinical and clinical phases. In the post-marketing phase, feedback from the “real world” of the clinic generates new observations, which lead to further testable hypotheses, and the cycle continues. Thus clinical research begins and ends with humans in our environment
1.2 Imhotep
Imhotep lived around 3000 BC. A polymath, Imhotep was an astronomer, conjurer, priest, architect, dentist, surgeon, and pharmacobotanist. While debated, he is believed to be the author of the Edwin Smith Papyrus. In it, there are 48 detailed clinical case studies, demarcated by organ system. The case reports are written like modern day SOAP notes. Each case is given a title, which suggests the presenting nature of the problem. The title is followed by an inspection and examination of the patient. This is followed by a diagnosis, prognosis, and treatment plan. The case studies provide detailed knowledge of organ function. It contains the first written reference to the brain. The relationship of brain and spinal injuries to paralysis are described, such as an understanding that crush injuries of the spine affect the body differently at each spinal level. The depth of medical knowledge and physiology in the papyrus suggested a sophistication that exceeded Hippocrates and preceded him by nearly 2500 years.
1.3 Shen Nong
Shen Nong (神 農, literally “divine farmer”) was a legendary Chinese Emperor who lived around 2700 BC. He is believed to be the author of the Shen Nong Ben Cao Jing (神農本草經) a book on medicinal plants and tea [1, 2]. It consists of three volumes. The first describes the beneficial effects of nontoxic stimulants such as ginseng, orange, and cinnamon. The second volume covers extracts used to treat human disease such as cucumber and ginger. These are listed as mildly toxic. The third volume catalogs toxic substances such as those derived from peach pits and rhubarb.
1.4 Hippocrates
Hippocrates of Kos (400 BC) promoted medical ethics and professionalism through his Oath [3]. He was a proponent of natural (as opposed to supernatural or divine) causes of disease. He developed a theory of medicine that disease was not a punishment for sin but caused by environmental factors or lifestyle. He contributed 42 case reports to the medical literature. He described dermatologic phenomena such as clubbing. He was the first to use the Greek word Karkinos to describe the crab-like extensions of blood vessels to a central bulbous tumor mass.
1.5 Sushutra
A near contemporary of Hippocrates, Sushruta (600 BC) of Varanasi wrote a surgical text, the Sushruta Samhita (सुश्रुतसंहिता), which described detailed skin surgery, plastic surgery, and reconstructive surgery including rotation flaps and pedicle flaps. To maximize training and minimize harm to patients Sushruta Samhita outlines meticulous practice of procedures on vegetables, plants, bamboo, animal skin, and dead animals.
1.6 Galen
Galen (Γαληνός, meaning “calm”) of Pergamon (150 BC), a patrician’s son, was a philosopher and physician to emperors Marcus Aurelius, Commodus, and Septimius Severus [4]. One of the most prolific authors of medicine and philosophy in ancient Greece, he is believed to have written or dictated nearly ten million words, a third of which survive. He has written major texts on physiology, anatomy, pharmacology, and diseases. He described several important diseases, such as the Antonine Plague (likely smallpox). He conducted animal experiments to understand human disease. He performed dissections on living and dead animals. He studied the four humors (blood, phlegm, yellow bile, and black bile) of Hippocrates’ time. In animal studies, he made the distinction between venous (dark red) and arterial (bright red) circulation. In a form of Imhotep redux, he transected the spinal cords of animals to show paralysis and inferred his findings applied to human disease.
1.7 The Middle Ages
Many of the Galenic texts were translated into Arabic and contributed to the rise of Islamic medicine during the Dark Ages and early Middle Ages. During this period, pharmaceuticals such as opiates were commonly found along trade routes. Animal studies were done. The beginnings of the understanding of anatomy and physiology were developed through animal studies and ultimately work on human cadavers. Theories of disease went from supernatural to natural. Experiments done on human subjects involved controls. The concept of consent was developed.
1.8 Avicenna
Avicenna (سينا بن) of Bukhara (1000 AD) wrote the Canon of Medicine (الطبالقانون في) In it, he expands on the work of Galen and describes investigational pharmaceutical principles which hold to this day, including use of pure drug, dose escalation, control groups, reproducibility, confirmation of animal tests in human subjects, and long-term observation [5].
1.9 Circulation
In the seventeenth century, blood and the circulation became better understood, through the work of William Harvey—who described circulation through the heart, lungs, body, and back—and Richard Lower and Edmund King who performed early blood transfusions. In Galen’s time, the blood was believed to go from the left side of the heart to the right through small pores or perforations in the septum. Harvey and his contemporaries were able to show the pulmonary circulation as the bridge between the right and left ventricles.
1.10 Scurvy
James Lind, 1747 AD, developed the concept of a control group. He performed a study of scurvy, dividing 12 sailors into 6 groups of pairs [6]. The pairs who were given cider, seawater, vinegar, sulfuric acid, or a mixture of nutmeg/garlic/horseradish did not improve. Only the group given one lemon daily improved. Scholars have since critiqued the lack of informed consent in Lind’s study.
1.11 Communicable Disease
In the 1700s, Edward Jenner developed a method for smallpox vaccination [7, 8]. Vaccination played an important role in the protection of George Washington’s troops during the Revolutionary War. Laws were passed to ensure the purity of vaccines and the qualifications of those administering vaccines [8–10].
In the eighteenth century, a case in which two surgeons disunited a partially healed fracture, lead to one of the early requirements for informed consent (1767, Slater vs. Baker & Stapleton). John Snow, an anesthesiologist, showed cholera was spread by the water supply, and is credited with founding epidemiology as a discipline.
Oliver Wendell Holmes, in 1855, was the first to notice that puerpural sepsis was contagious and likely caused by transmission from physicians conducting autopsies on cases of puerperal fever. Ignaz Semmelweis conducted a clinical trial on hand washing in the prevention of puerpural fever in the maternity ward of Vienna General Hospital. The results of this and another trial were published in 1861. These findings, along with those of Koch, Pasteur, and Lister ushered in an era of bacteriology and the study of infectious disease. In 1874, Gerhard Armauer Hansen, deduced from epidemiologic studies that M. leprae is the cause of leprosy. To overcome his critics, in 1880 he inoculated patients and nurses with the bacillus to show causality. This was done without consent. Walter Reed, in studying the mosquito as a vector for yellow fever, obtained informed consent from all his volunteers, and noted as much in all his publications.
1.12 Antibiotic Era (Fig. 1.2)
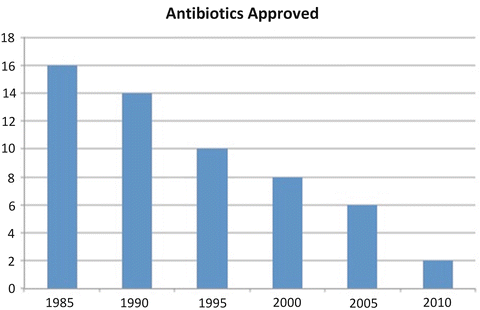
Fig. 1.2
The number of new antibiotics approved by the FDA has declined precipitously. Source: H. Boucher Pew Charitable Trust Meeting
In 1925, Abraham Flexner issued a report requiring a rigorous scientific basis for medical education. In 1928, Scottish physician Sir Alexander Fleming, discovered penicillin, and essentially gave birth to a pharmaceutical industry, beginning with antibiotics. In the 1930s, the first generation of antibiotics was discovered. This included the beta lactams, sulfa drugs, the aminoglycosides, and chloramphenicol. In the 1950s tetracyclines, macrolides, and quinolones were developed [11]. For the next two decades, antibiotic research languished and any subsequent antibiotic advances were in the form of incremental, so-called “me too” drugs. Some analysts have blamed this drought on the FDA’s statistical requirements for proving noninferiority. The FDA relaxed their requirements in a meeting with PhRMA and IDSA (Infectious Diseases Society of America), however relations between the FDA and industry reached another low point in 2006 in the wake of the telithromycin (Ketek) trial, which resulted in withdrawal of approval of a drug that caused rare but serious liver toxicity. Most companies have withdrawn because:
Clinical trials for antibiotics are becoming more expensive because more targets of efficacy (species of organism, and site in the body) are required to show noninferiority over competitors.
Companies are more interested in chronic diseases. Antibiotics are for short-term use only. The net present value (NPV) of a drug represents its lifetime earnings minus its lifetime costs. The NPV of antibiotics has been $1.1 B compared to $11 B for SSRIs (selective serotonin reuptake inhibitors) and $15 B for statins.
Resistance makes products less effective and less profitable. Stronger agents are held in reserve in small stockpiles in a few hospitals, also reducing profitability for companies.
The need for antibiotics could not be greater, with resistance increasing, and with the emergence of the so-called ESKAPE pathogens (Enterococcus, Staph, Klebsiella, Acinetobacter, Pseudomonas, Enterobacter). This family of pathogens is responsible for a growing number of serious infections of the skin and other organs and has few effective treatments [12, 13]. If regulatory costs prohibit advances in treating these infections (for example, nanoparticle trapped nitric oxide), manufacturers may develop and market the next generation of antibiotics outside the US. To reduce costs and promote innovation, some companies are banding together. Bristol-Meyers Squibb and Gilead Sciences came together on making a combination HIV pill. Merck published the crystal structure of HIV protease for competitors to use.
1.13 Industry and Regulation
During World War II and after, pharmaceutical research became a large enterprise sponsored by government and industry. Large numbers of trials were conducted on captive volunteers, such as military personnel, prisoners, and institutionalized individuals (mentally ill, orphans, physically handicapped). In fact, many large academic medical centers and pharmaceutical companies had their research sites located near institutions, sometimes just across the street. Mishaps, tragedies, and cases of wartime and peacetime abuse led to ethics convocations and the promulgation of laws protecting human subjects and empowering agencies such as the FDA to develop guidelines to ensure the safety and ethical conduct of clinical research involving human subjects.
The US began receiving counterfeit and ineffective drugs from Mexico, leading to the Import Drugs Act of 1848. In 1905 Upton Sinclair’s “The Jungle” exposed unsanitary conditions in the Chicago meatpacking industry. This instigated legislation requires processing inspections, and forbidding interstate and foreign commerce in impure and mislabeled foods and drugs.
The Food and Drug Act of 1906 did not require drugs to be effective, just that they meet standards of strength and purity. Before the act, drugs were commodities, and their contents were secret, hence the name patent medicine. In 1938, after 107 deaths due to “Elixir Sulfanilamide” the FDA required manufacturers to prove drug safety before marketing.
In 1947, the Nuremberg Code required informed consent prior to participating in experiments. In 1962, the Estes Kefauver-Harris Amendment led to requirements of teratogenicity and reproductive effects of drugs to be added, following the thalidomide phocomelia epidemic. During the Bay of Pigs in 1961, 1,200 men were captured, and ransomed from Cuba for $50M of drugs and supplies donated by the US Pharma. In return, they got tax deductions, and political good will. This resulted in the Drug Abuse Control Amendment of 1965, making it a crime to infringe on drug copyright or branding, leading to penalties, seizures of assets, or imprisonment.
In 1982 the maker of Tylenol recalled 31 million bottles, valued at $100 M, and developed tamper-proof bottles. The Anti-Tampering Act passed in 1983, requires tamper-resistant packaging, and makes tampering a crime. The focus of the FDA has evolved over the last few decades: 1970s– 80s randomization and blinding; 1990s metabolism; 2000s safety (suicides on antidepressants, statin myopathy, aprotinin and surgical blood loss, Cox-2 and heart attacks, efalizumab and progressive multifocal leucoencephalopathy, topical immunomodulators and skin cancer risk, tacrolimus and Netherton syndrome) led to the FDA’s 2007 Amendments Act, with an emphasis on risk mitigation and pharmacovigilance. Now the emphasis seems to be on comparative effectiveness research.
1.14 Protocol Design
Sponsors typically provide sites with an identical protocol which cannot be modified. Some sponsors will reserve funds for small investigator-initiated studies, especially if they want to create good will at the site. Usually, these are conducted at large academic medical centers, with funding through grants, which are written by the investigators.
Inset 1.1
Nanotechnology clinical trials in melanoma. Matter behaves differently on the nanoscale. Its behavior depends upon its size, surface potential, surface reactivity, surface-to-volume ratio, shape, and other properties. One recent way to augment cell-mediated immunity has been to couple antigens to nanoparticles and immunostimulatory compounds. A small Phase-I trial in Europe examined the effect of a nanoparticulate vaccine combined with Toll-like receptor agonists on memory cells in melanoma subjects.
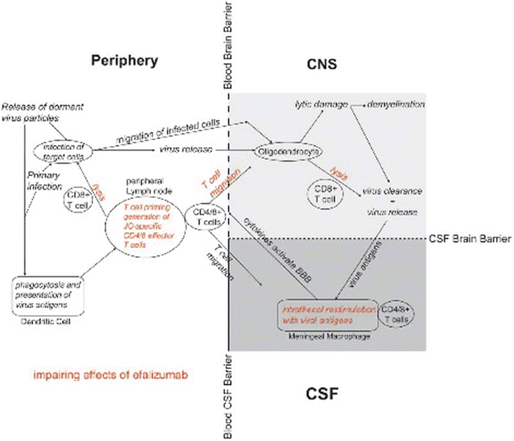
Goldinger SM, Dummer R, Baumgaertner P, Mihic-Probst D, Schwarz K, Hammann-Haenni A, Willers J, Geldhof C, Prior JO, Kündig TM, Michielin O, Bachmann MF, Speiser DE. Nanoparticle vaccination combined with TLR-7 and -9 ligands triggers memory and effector CD8+ T-cell responses in melanoma patients. Eur J Immunol. 2012 Nov;42(11):3049–61.
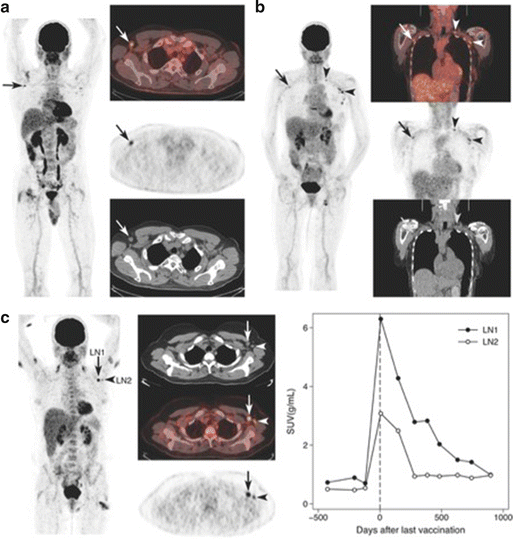
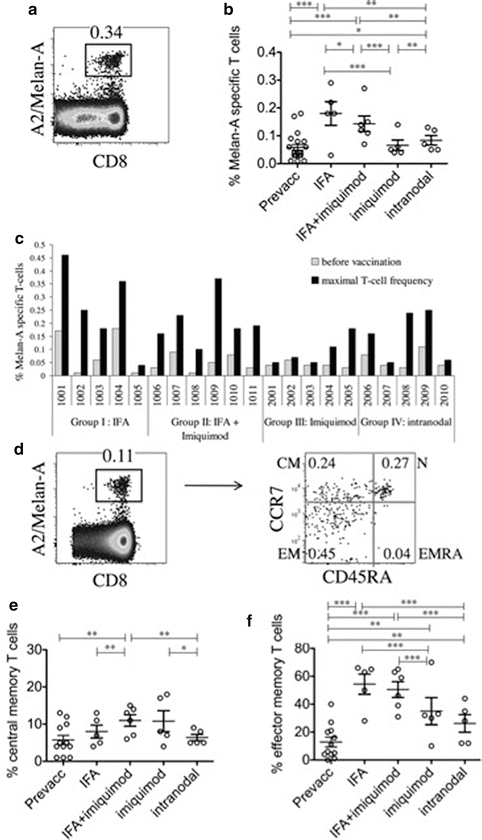
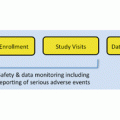
When you undertake a clinical investigation, your guide is a protocol. Whether you write your own protocol for an investigational new drug or device, or obtain a protocol from a sponsor, it contains stereotypical elements. Typical protocol components include:
Introduction: This section explains the illness, and the rationale for the intended drug or device.
Objectives: These depend on the phase of the study, early phase objective might be tolerability, while later phase objectives might be safety and efficacy.
Plan: This section discusses the details of the study. The size of the study, the targeted treatment populations, and the arms or study treatment groups are described here.
Inclusion/exclusion criteria spell out in as much detail as possible who may or may not participate in the study.
Methodology: This is a step-by-step guide to each study visit. Elements include instructions for examining subjects, photographing skin lesions, taking biopsies, administering medication, and entering data in an electronic case report form. This section should be written clearly enough and in sufficient detail that any outside person could reproduce the study.
Termination criteria: This covers end points, such as improvement in Psorasis Area and Severity Index (PASI) score by 10 %. This section often requires a statistician’s help.
Adverse events: This section has a clear definition of adverse events and severe adverse events as well as clear reporting guidelines and reporting timelines.
Laboratory procedures: Covers special tests that the study may require, such as venipuncture, or electrocardiogram, biopsy, or imaging.
Administrative: This section details the administrative responsibilities of the site, the sponsor, any contracting group such as a CRO or SMO, and any regulatory agency such as an IRB or the FDA.
Statistical planRelated posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree