Fig. 13.1
Contrasting conventional and alternative models for why cancer incidence increases with age. (a) Illustrates the conventional dogma that suggests oncogenic mutations create “super cells” that have surpassed the fitness optimum leading to oncogenesis. (b) The Adaptive Oncogenesis Hypothesis suggests that the incidence of cancer is not just dependent on the accumulation of mutations, but the ability of these mutations to make the harboring cell more “fit” relative to other cells (competitors) in the niche. This model suggests that mutations are typically maladaptive in healthy backgrounds due to their inability to improve optimal fitness levels; however, in damaged pools (such as in aged backgrounds), these mutations improve cellular fitness leading to increased selection for oncogenically initiated cells. Fitness is defined as the ability of cells of a specific genotype or epigenotype to contribute to current and subsequent cell generations due to their phenotypic properties (proliferative capacity, survival capabilities, maintenance of stemness, etc.)
Therefore, after summarizing what is currently known about HSC aging and malignancies associated with this process, we will discuss current attempts to explain the aging-dependent mechanism of carcinogenesis. It is clear that both aging hematopoiesis and aging-associated hematopoietic malignancies will be multifactorial. We will also describe an alternative model (adaptive oncogenesis), which suggests that fitness effects of oncogenic mutations are context dependent, generally conferring disadvantages to incipient cells in highly fit pools but gaining selective advantages within aged/damaged stem cell pools in the aged tissue microenvironment. This model predicts that aging alters the general adaptive landscape for stem cell pools, which promotes increased selection for oncogenically initiated cells.
This chapter will address a number of open questions in understanding age-dependent changes in hematopoiesis and increases in leukemia incidence, such as:
Why does the HSC pool become more biased towards the production of myeloid cells with increasing age, and what is the relationship between this changing HSC bias and declining HSC fitness with age?
How does this shift in HSC bias and HSC fitness with age influence leukemogenesis?
How do other changes in hematopoiesis with age influence leukemogenesis?
Given that leukemias associated with aging, such as acute myeloid leukemia (AML), chronic myeloid leukemia (CML), and chronic lymphocytic leukemia (CLL), are thought to have HSC origins, what changes in HSC with age could account for the dramatically increased incidence of these leukemias in old age?
13.2 Aging-Associated Changes in Hematopoietic Development and Malignancies
The proportion of elderly people is progressively rising throughout the world, with the most pronounced increase in the developed world. Elderly people (>65 years old) are expected to comprise greater than 20 % of the global population by 2050 (Dorshkind et al. 2009). This increase is posing major challenges for healthcare systems, as aging is associated with marked increases in a number of diseases, including most types of cancers (Rossi et al. 2007a; Benz and Yau 2008; Blagosklonny et al. 2010). With more than 80 % of human cancers being diagnosed after the age of 50 (DePinho 2000), aging represents the single most important prognostic factor for many cancers, including lung, colon, prostate, and certain leukemias (Benz and Yau 2008; Edwards et al. 2002; Balducci and Beghe 2001). Strong associations between aging and cancer are traditionally used to support the mutation-centric view of clonal evolution of cancers: aging leads to an accumulation of random mutations, and since (a) some of these random mutations are expected to activate cellular oncogenes or silence suppressor genes and (b) transformation is thought to require cooperation between several oncogenic events, aging should translate into increased risk of cancer initiation (Serrano and Blasco 2007). On the other hand, aging is also associated with substantial cell-autonomous and microenvironmental changes (Henry et al. 2011). In principle, these changes are likely to modify the ability of oncogenic mutations to drive clonal expansion. In fact, some of the age-related changes such as increased inflammation and decreased immune surveillance have been clearly implicated in carcinogenesis (Henry et al. 2011). In addition, intracellular changes such as telomere shortening as well as growth inhibitory changes in the microenvironment could create a scenario for a strong selection for “oncogenic resistance to growth inhibitory conditions” (Blagosklonny 2002). Thus, the effects of aging are highly complex and span organismal, systemic, and cellular levels (Anisimov 2009). Cancer development is similarly complex. Thus it would be naïve to expect a simple relationship between mutational accumulation and the increasing incidence of aging-associated cancers.
One of the more notable age-related changes in hematopoiesis is immunologic decline (Kirkwood 2005; Henry et al. 2010, 2011; Anisimov 2009; Greaves and Maley 2012; Greaves and Wiemels 2003; Naumov et al. 2006a), which impairs responses to pathogens and reduces vaccination efficacy in elderly populations. Decreased immune function is not compartmentalized; reduced immune cell function (and in some cases cell numbers) has been observed in both the myeloid and lymphoid lineages (Weiskopf et al. 2009; Weinberger et al. 2008; Linton and Dorshkind 2004). Furthermore, recent studies have indicated that these functional reductions result at least in part from aging-associated defects in HSC function, which are transferred to their lineage-committed progeny (Weiskopf et al. 2009; Linton and Dorshkind 2004). The causes of the aging-associated decline in HSC and hematopoietic cell function are still under investigation.
13.2.1 Aging and HSC Fitness
Most of our current knowledge on the impact of aging on HSC function comes from studies using mouse models (Weiskopf et al. 2009; Linton and Dorshkind 2004; Sharpless and DePinho 2007; Miller and Allman 2005). These studies implicate both cell-intrinsic and cell-extrinsic factors towards impairments of age-dependent alterations in HSC behavior, although within animals both intrinsic and extrinsic influences are tightly interwoven and are sometimes difficult to disentangle (Chambers and Goodell 2007). Extrinsic factors, including changes in niche composition and hormone production, have been postulated to play a major role in declining HSC function with age (Chambers and Goodell 2007; Yin and Li 2010). In vivo, multiphoton intravital microscopic analysis of HSC in the bone marrow of young and old mice has revealed that old HSC resides further away from the endosteum than young HSC progenitors, indicating age-related extrinsic changes in niche composition that could impact HSC function (Kohler et al. 2009). HSC normally resides in the hypoxic endosteum microenvironment (Kohler et al. 2009; Simsek et al. 2010; Takubo et al. 2010). The effects of hypoxia on HSC function are just beginning to be delineated. It has been postulated that hypoxia regulates HSC self-renewal by limiting cell cycle entry (increasing self-renewal), which would limit potential accumulation of damage (Takubo et al. 2010; Eliasson et al. 2010). Thus, residing further away from the endosteum, by increasing oxidative damage to DNA, could increase the mutational load in HSC (although this has yet to be demonstrated). In addition, it is believed that hypoxia limits the flow of extracellular fluids into the endosteum niche, which reduces HSC exposure to potentially hazardous toxins and proinflammatory cytokines, some of which are known to be tumor promoting (Takubo et al. 2010; Eliasson et al. 2010; Karin et al. 2006; Rossi et al. 2007b). Indeed, aging in mice is associated with increased detection of γH2AX foci in HSCs (Rossi et al. 2007b), which could reflect DNA damage. Notably, a recent study showed that these foci reflect increased replicative stress in old HSCs, coinciding with decreased expression of components of the replication complex (MCMs) (Flach et al. 2014). Should mutation load increase in HSCs in old age, the chances for acquisition of oncogenic mutations would be increased, which could contribute to the increased initiation and progression of cancers in the elderly (whether mouse or human). Moreover, another consequence of increasing replicative stress and mutational load in HSCs (as in other stem cell types) could be the reduction in stem cell fitness, as DNA damage is more likely to negatively impact on cellular functions than improve them. Beyond the accumulation of genetic damage, epigenetic drift with age also likely contributes to HSC fitness decline (Chambers et al. 2007; Wahlestedt et al. 2013; Bell and Spector 2011; Sun et al. 2014). Moreover, other cell-autonomous changes likely contribute. Indeed, HSCs in old mice exhibit greater dependence on autophagy, which is selectively needed for old HSCs to maintain cellular energetics and survival (Warr et al. 2013). Notably, recent computational studies of changes in HSC fitness with age indicate that cell-autonomous accumulation of genetic and epigenetic events is insufficient to account for fitness decline with age, instead revealing that microenvironmental alterations with age are required to achieve the observed severalfold reductions in HSC fitness in old age (Rozhok et al. 2014).
Aging-related, cell-autonomous changes have also been shown to contribute to altered HSC function (Chambers et al. 2007; Rossi et al. 2005; Chambers and Goodell 2007; Warren and Rossi 2009; Beerman et al. 2010a, b). When highly purified long-term HSCs (LT-HSCs) from young or old mice are used to reconstitute young irradiated recipient mice, old LT-HSCs are two- to fourfold less efficient at reconstitution per HSC, and their production is skewed in favor of myelopoiesis, with accompanying reductions in lymphopoiesis (Dykstra et al. 2011; Chambers et al. 2007; Rossi et al. 2005; Henry et al. 2010; Miller and Allman 2005; Chambers and Goodell 2007; Morrison et al. 1996). Thus, as defined above, old HSCs are less “fit,” in that young competitors are better able to contribute to hematopoiesis. Aged mouse HSCs also exhibit decreased homing and engraftment posttransplantation (Morrison et al. 1996; Liang et al. 2005).
The hematopoietic system may attempt to compensate for decreased HSC function (and decreased production rates of mature progeny) by increasing the size of the HSC compartment, as indicated by the observation that the number of phenotypic HSCs in C57Bl/6 mice increases with age (Chambers et al. 2007; Rossi et al. 2005; Chambers and Goodell 2007; Morrison et al. 1996; Sudo et al. 2000; de Haan and Van Zant 1999; Noda et al. 2009). Such increased homeostatic mechanisms, both through increased cytokine levels and perhaps increased turnover rates for HSCs and other progenitors, should contribute to higher chances for mutation fixation. Larger numbers of HSCs should also increase the target size for oncogenic mutations. However, increased HSC numbers have not been observed in all mouse strains; BALB/c and DBA/2 do not show an age-dependent increase in the numbers of phenotypic HSC (Van Zant et al. 1990; Chen et al. 1999, 2000). Still, increased numbers of HSCs, at least as assessed using phenotypic markers, are observed in elderly humans (Pang et al. 2011). Although the effects of aging on human HSCs have not been as extensively studied, human HSC function also appears to be affected by aging given the observation that the proliferative potential of human HSCs declines with age (Vaziri et al. 1994). In addition, bone marrow from older humans is less efficient at reconstituting recipients when compared to the reconstitution capacity of bone marrow derived from younger patients (Kollman et al. 2001). Therefore, based on experiments in mice and more limited observations in humans, it is clear that a number of age-associated alterations in the HSC compartment, due both to extrinsic and intrinsic factors, could contribute to increased frequencies of hematopoietic malignancies in the elderly.
13.2.2 HSC Lineage Bias Changes with Age
Aging-associated changes in HSC reflect more than just a decline in fitness. In aged mice, it has been demonstrated that a developmental shift occurs, with reduced lymphopoiesis in favor of greater myelopoiesis. Microarray analyses of HSCs isolated from old mice reveal a gene expression profile consistent with this age-dependent bias towards production of myeloid cells (Chambers et al. 2007; Rossi et al. 2005; Chambers and Goodell 2007). The myeloid bias appears to be at least in part cell autonomous, as adoptive transfer of young and old HSC into young, irradiated recipients recapitulates the age-dependent bias towards myelopoiesis (Rossi et al. 2005; Dorshkind et al. 2009; Guerrettaz et al. 2008). This bias appears to be mediated by a shift towards greater frequency of myeloid-biased (mb-HSCs) relative to lymphoid-biased HSCs (lb-HSCs) (Sudo et al. 2000; Muller-Sieburg et al. 2004; Cho et al. 2008), and these differentially biased HSCs can now be prospectively isolated and shown to transfer their developmental bias to recipient mice (Beerman et al. 2010a, b; Challen et al. 2010; Kent et al. 2009). Notably, transplantation of mb-HSCs from young and old mice at limiting dilution (so that a single HSC contributes to hematopoiesis in the recipient) reveals a significant two- to threefold reduction in engraftment for each successfully engrafting old mb-HSC relative to young mb-HSC (indicating a per cell reduction in HSC fitness in old mice, even when similarly “biased” HSCs are compared) (Dykstra et al. 2011). It is important to note that while myelopoiesis becomes favored over lymphopoiesis in old mice, myeloid progenitors are not “more fit” relative to the young (Dorshkind et al. 2009; Signer et al. 2007). Moreover, elderly humans exhibit alterations in HSC representation and decreased developmental potential of both lymphoid and myeloid progenitors (Pang et al. 2011; Sharpless and DePinho 2007; Kuranda et al. 2011). Notably, human BM cells with HSC phenotypic markers also show myeloid skewing and decreased repopulation potential in immunocompromised mice, suggesting decreased fitness (Pang et al. 2011).
Recently, it has been demonstrated that increases in the activation of the noncanonical Wnt signaling pathways, mediated primarily through Wnt5a, are an important contributor to HSC aging (Florian et al. 2013). Specifically, aging promotes increased expression of Wnt5a in long-term HSCs which leads to decreased β-catenin activity in the nucleus (Florian et al. 2013). Furthermore, Wnt5a treatment of young HSC induces an aging-like phenotype, indicated by increases in Cdc42 levels, decreases in the percentage of polarized LT-HSCs, and reductions in B lymphopoiesis with accompanying increases in myelopoiesis in vivo (all of which are phenotypes observed in aged HSC (Florian et al. 2012)). Additionally, aged LT-HSCs isolated from Wnt5a-haploinsufficient mice have reduced Wnt5a and Cdc42 activity, corresponding with better maintenance of B lymphopoiesis without accompanying increases in myelopoiesis in vivo (Florian et al. 2013). Transplantation studies revealed that reducing Wnt5a activity in aged LT-HSCs reversed the aging-associated, cell-intrinsic deficiencies exhibited by a significant increase in the capacity of these cells to make B cells. As a result of the findings from this study, we now have a better understanding of the molecular processes that underlies LT-HSC aging and how this shift from canonical to noncanonical Wnt signaling pathways promotes aging hematopoiesis. Importantly, these studies suggest that at least some aspects of the aging hematopoietic phenotype are reversible by altering signaling, which could also provide insight into how particular oncogenic events may be adaptive in aged HSC pools.
13.2.3 The Impact of Aging on Innate and Adaptive Immunity
The biological consequences of aging-associated alterations in hematopoiesis and the impact on the ability of mature immune cells to control aging-associated cancers are being actively investigated. It has been extensively shown that innate and adaptive immune cells from older individuals exhibit reduced function and/or numbers, which decrease their ability to eliminate aging-associated cancers. Decreased cellular function in old age has been documented in innate cells such as dendritic cells (DC) (reduced T-cell stimulatory capacity) and natural killer (NK) cells (reduced cytolytic potential) (Weiskopf et al. 2009; Shaw et al. 2010). Age-associated impairment of the adaptive immune system, composed of T cells and B cells, is also observed in both aged mice and humans (Dorshkind et al. 2009; Miller and Allman 2005; Dorshkind and Swain 2009; Allman and Miller 2005) and could facilitate cancer development in older individuals. In mice, the production of naïve CD4+ T cells (T-helper cells) declines with age (Aspinall and Andrew 2000a, b). CD8+ T-cell function, as measured by decreased proliferation in response to IL-2 and reduced expansion and differentiation upon antigenic stimulation, also declines with age (Weiskopf et al. 2009). Aging B lymphopoiesis in mice is characterized by reductions in progenitor cell populations, reduced responsiveness to growth factors such as interleukin-7 (IL-7), and decreases in their ability to undergo class switching. In humans, the aging-associated decline in B-cell lymphopoiesis appears less drastic compared to mice, and it has been demonstrated that B-cell lymphopoiesis in humans is less dependent on IL-7 stimulation (Rawlings et al. 1995; Prieyl and LeBien 1996). The decline in humoral responses in elderly people is largely attributed to changes in the composition of the cells comprising the B-cell repertoire (Weiskopf et al. 2009; Weksler et al. 2002; Weksler and Szabo 2000). Although the effects of aging on B lymphopoiesis differ between mice and humans, there is significant overlap in the decline in B-cell function found in aged population from both species. While reductions in B lymphopoiesis in old age could contribute to reduced tumor immune surveillance, there has been more debate about the implications of perturbed B-cell development towards leukemogenesis (described below).
13.2.4 Inflamm-aging and the HSC Repertoire
Aging-associated changes in HSC representation and function, some of which are subsequently imprinted on mature immune cells, prompt two important questions: (1) What factor(s) induces the aging-associated alterations in HSCs? (2) What is the biological cause (and consequence) of these changes? Regarding the former, increases in aging-associated inflammation, dubbed “inflamm-aging,” may impinge on stem cell populations leading to the altered stem cell dynamics discussed in the previous sections. Indeed, inflammatory cytokines have been shown to regulate HSC activity with cytokines such as TGF-β and TNF-α showing the capacity to promote quiescence by repressing HSC expansion (Pronk et al. 2011; Yamazaki et al. 2009; Mirantes et al. 2014; Pietras et al. 2014), while the cycling of HSCs can be induced through the actions of IFN-α and IFN-γ (Essers et al. 2009; Baldridge et al. 2010). Furthermore, recent evidence suggests that HSC skewing towards the myeloid lineage in old age may be in part promoted by cytokines like TGF-β1 (Chambers et al. 2007; Challen et al. 2010; Song et al. 2010) or by perturbation of the bone marrow microenvironment (Omatsu et al. 2010). “Inflamm-aging” is characterized by increased levels of tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β) in the plasma (Weiskopf et al. 2009; Franceschi et al. 2000). This increase in the circulating levels of inflammatory cytokines likely reflects cell-autonomous changes in myeloid gene expression resulting in microenvironmental alterations that could favor tumor initiation and progression (Weiskopf et al. 2009). In addition, aged HSCs themselves have been shown to exhibit proinflammatory and stress response biases in their gene expression profiles (Chambers et al. 2007). The cause of this aging-associated increase in inflammation is still under investigation. However, proposed culprits of “inflamm-aging” are chronic infection by pathogens past reproductive ages, elevated aging-associated stress responses, declines in mechanisms responsible for dealing with these stresses, and altered cellular metabolism (Chambers et al. 2007; Simsek et al. 2010; Franceschi et al. 2000; Garaycoechea et al. 2012). In fact, Franceschi et al. propose that humans (and presumably other mammals) evolved to deal with chronic infections during our “normally” short lives (at least for most of our evolutionary history) (Franceschi et al. 2000). These same mechanisms, which were beneficial for controlling infections through reproductive years, could be contributing to increased inflammation in old age (of course, natural selection acted on the former to a far greater extent than the latter).
Chronic inflammation is a well-defined contributor to tumorigenesis, with tumor-associated macrophages (TAMs) playing a key role in driving tumorigenesis (Franceschi et al. 2000). The cytokines produced by these TAMs, such as TNF-α, could influence cancer progression both by promoting proliferation of oncogenically initiated or more advanced tumor cells and by inducing death of nonmalignant stem and progenitor cells (thus stimulating compensatory proliferation) (Karin et al. 2006; Karin and Greten 2005). Inflammation may also create a hazardous microenvironment, with increased ROS and cell death, thus promoting selection for oncogenic events that confer resistance or are otherwise adaptive to this altered microenvironment.
On the HSC level, inflammation likely induces changes in the microenvironment that increases exposure of HSCs to insults that could induce damage and subsequently lead to decreased fitness (Chambers et al. 2007; King and Goodell 2011). As mentioned above, HSCs in the bone marrow of old mice reside further away from the protective hypoxic endosteum than their young counterparts (Kohler et al. 2009). This altered localization correlates with increased detection of γH2AX foci and loss of quiescence (mediated by reduced HIF-1α levels) (Takubo et al. 2010), which may result from elevated exposure to toxins and cytokines (King and Goodell 2011). Furthermore, loss of HIF-1α has been shown to alter HSC metabolism (Simsek et al. 2010), which could alter cellular fitness. Thus, aging-associated inflammation may also promote cell-intrinsic (whether genetic or epigenetic) and cell-extrinsic changes, which alter cellular fitness, subsequently leading to increased selection for oncogenically initiated cells.
Regarding the latter, why does the representation and fitness of HSC change with age? As discussed previously, hematopoietic aging in mice and humans is accompanied by a skewing towards myelopoiesis. This shift is attributed at least in good measure to changes in the genetic program of HSC and a reduction in the number of lymphoid-biased HSC. From an evolutionary perspective, this shift in the representation of hematopoietic progenitors appears programmed, since the majority of the lymphocyte repertoire is established during youth. Indeed, T lymphopoiesis declines starting before full maturity (Dorshkind et al. 2009; Dorshkind and Swain 2009) and thus clearly represents an evolved program (not a consequence of decreased maintenance in old age). Similarly, B lymphopoiesis declines gradually with age (Miller and Allman 2005; Milne and Paige 2006), similarly suggesting that this programmed switch in hematopoiesis (to more myeloid) provides an adaptive advantage to mammals. Thus, these changes in hematopoiesis are advantageous for organismal fitness: the lymphocytic repertoire is established relatively early in life and can be subsequently maintained at the mature T- and B-cell levels. In contrast, a constant supply of myeloid cells, which do not self-renew and represent the first line of defense against pathogens (in addition to playing many other roles in tissue maintenance) (Cain et al. 2009), is required throughout life.
Notably, not all aging-associated changes are cell intrinsic. Several reports show that microenvironmental alterations that occur during aging alter HSC function. These include the reduced ability of old bone marrow stromal cells to support hematopoietic progenitor cell expansion, reduced adhesion of aged HSC and progenitor cells to bone marrow stroma, and reduced polarity of aged HSC to BM stromal cells (Florian et al. 2012; Stephan et al. 1998; Xing et al. 2006). Moreover, inflammation has been shown to directly impair B lymphopoiesis (and thus favor myelopoiesis), by preventing B-progenitor localization to the IL-7-rich niche (Ueda et al. 2005). While inflammation is important for animal survival in youth (for eliminating infections and repairing tissues), it can become detrimental in old age in its chronic manifestation (Goto 2008). These observations suggest that microenvironmental stimuli play a role in the regulation of stem and progenitor cells and the subsequent skewing that accompanies aging. Thus, while some myeloid skewing could be the result of the programs described above, additional skewing towards myelopoiesis could result from age-associated inflammation (at ages beyond the point where natural selection has much influence).
Taken together, these data suggest that aging hematopoiesis potentially results from two phenomena: (1) aging-associated decline in lymphoid-biased HSC populations resulting from a program favored by natural selection (B- and T-cell repertoires are established early, allowing hematopoiesis to focus on myeloid cell production, as myeloid cells are the first line of defense against pathogens) and (2) aging-associated changes in HSC that result truly in old age (beyond much influence from natural selection, such as due to inflammation), which decrease the fitness of HSC and subsequently progenitor cells (Fig. 13.2).
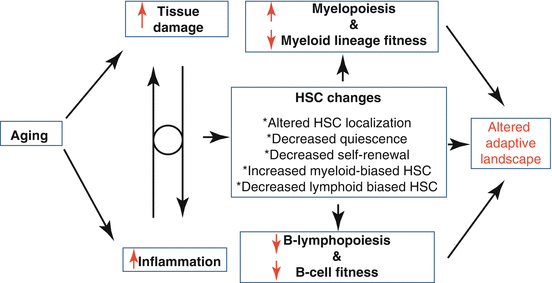
Fig. 13.2
Model for aging-associated changes in the adaptive landscape. Aging is accompanied by increasing tissue damage and elevated levels of inflammation “inflamm-aging.” We postulate that these events modulate HSC function by promoting localization away from the protective hypoxic endosteal niches. Aging HSCs also exhibit cell-intrinsic alterations which lead to decreased quiescence, reduced self-renewal, and altered differentiation, favoring the expansion of myeloid-biased HSCs while suppressing the maintenance of lymphoid-biased HSCs. Overall, these age-dependent alterations of the HSC pool should result in fitness declines in stem and progenitor cells ultimately resulting in drastic changes in the adaptive landscape
13.2.5 Aging, HSC, and Leukemogenesis: A Complicated Relationship
So how might aging-associated changes in HSC contribute to increased leukemogenesis? Stochastic aging-associated changes, such as inflamm-aging, could stress HSCs in older individuals. For example, elevated levels of proinflammatory cytokines such as TNF-α (which is frequently increased in the serum of older individuals) have been shown to increase cycling of mb-HSCs (Ueda et al. 2004). Chronic inflammation has also been associated with decreased HSC self-renewal, which is attributed to persistent mitochondrial ROS levels (Chambers et al. 2007; Goto 2008). Increased cycling of HSCs together with higher ROS levels (and subsequent damage to cellular macromolecules including DNA) could thus increase the chances for fixation of oncogenic mutations.
As we argued above, reducing the fitness of a cell population should increase selection for adaptive oncogenic events, thus promoting cancer development. Thus, an important question remains: how do changes in HSCs with age influence the fitness of committed progenitor cells? As described above, aging-associated defects in B and T lymphopoiesis are particularly apparent, which in mice can be attributed to both cell-intrinsic and cell-extrinsic effects of aging (Linton and Dorshkind 2004; Guerrettaz et al. 2008; Allman and Miller 2005; Miller and Allman 2003; Lescale et al. 2010). For T lymphopoiesis, age-dependent reductions (starting in youth) in T-cell production can be partially (but not entirely) ascribed to the involution of the thymus. B-cell progenitors exhibit clear cell-autonomous functional defects, such as reduced responsiveness to IL-7 (Miller and Allman 2003; Andrew and Aspinall 2001) and reduced kinase-dependent signaling (Henry et al. 2010). These functional defects have consequences, as we have shown that defective signaling and reduced fitness of old B-progenitor pools lead to potent selection for oncogenes like BCR–ABL that correct signaling defects (Henry et al. 2010). Moreover, stochastic modeling of somatic evolution in HSC pools revealed that non-cell-autonomous influences on HSC fitness through the microenvironment were necessary to explain the age dependence of leukemias (Rozhok et al. 2014).
But why would B-cell progenitors in old mice exhibit functional defects, as these progenitors are not themselves “old” (with relatively short life spans of a few weeks), but were derived from old HSCs? Notably, Muller-Sieburg et al. have shown that even young murine mb-HSCs (which dominate the hematopoietic system of old mice and humans) make lymphoid progenitors with reduced function compared to those derived from lymphoid-biased HSCs that are predominant during youth (Muller-Sieburg et al. 2004). Why would natural selection favor a mechanism whereby functionally impaired B progenitors are produced in young mice? In one speculative scenario, we propose that fit B lymphopoiesis is indeed maintained in young mice, as most B progenitors will be derived from lb-HSCs, and the few derived from mb-HSC will be outcompeted by those derived from lb-HSC (thus maintaining overall B-progenitor pool fitness). Indeed, Challen et al. showed that in competitive repopulation experiments, myeloid progenitor pools were dominated by progeny of mb-HSCs and lymphoid progenitor pools by progeny of lb-HSCs (Challen et al. 2010). Notably, this lineage bias was amplified by competition from progeny of HSCs with the opposing bias (i.e., lymphoid progenitors from mb-HSC appear to be outcompeted by lymphoid progenitors from lb-HSC) (Challen et al. 2010). Thus, only when mb-HSCs far outnumber lb-HSCs (in old animals, beyond the period of strong natural selection) would B progenitors actually be largely derived from mb-HSCs and thus exhibit reduced fitness.
Notably, impaired B lymphopoiesis in old age appears to be more than just mediated by changes in HSC subsets. Phenotypically distinct age-associated B cells (ABCs) have been shown to accumulate with advanced age in mice (Hao et al. 2011), and these ABCs exhibit heightened Toll-like receptor (TLR) responses (Hao et al. 2011) and accumulate in response to TLR signals (Rubtsov et al. 2011, 2013). ABCs from old mice also exhibit increased expression of TNFα, which can inhibit IL-7-dependent clonogenicity of pro-B cells (Frasca et al. 2012; Ratliff et al. 2010). Importantly, the Melamed lab has shown that depletion of mature B cells in old mice leads to restored B lymphopoiesis (Keren et al. 2011a, b).
So what are the implications for leukemogenesis? Beyond the obvious effects of reduced lymphopoiesis on immunity against cancers, the impaired fitness of lymphoid progenitors could promote selection for adaptive oncogenic mutations, as we have shown (Henry et al. 2010). While such oncogenic mutations might be expected to be adaptive in mb-HSC-derived B-progenitor pools (even if derived from young mb-HSCs), in a young animal B progenitors from lb-HSCs will dominate such pools (maintaining pool fitness and thus preventing oncogenic selection). On the other hand, the lack of lb-HSC-derived B lymphopoiesis in old animals could be responsible for the poor fitness of B-progenitor pools and consequent increased leukemogenesis. Finally, it is also interesting to consider how alterations in B lymphopoiesis mediated by mature B cells (such as ABC) might impact oncogenic adaptation and leukemogenesis in old mice. Importantly, whether mediated by changes at the level of HSCs or committed B progenitors, it will be important to determine whether modulation of inflammation can impinge on B lymphopoiesis and the adaptive landscape for oncogenesis.
If aging-associated mechanisms such as inflammation are indeed a major contributor to aging hematopoiesis, it stands to reason that if these insults could be reduced, it would lead to improvements in cellular fitness and reduced cancer incidence. Several observations support that this may be the case. First, it is notable that polymorphisms associated with reduced inflammation are preferentially found in centenarians (Franceschi et al. 2005; Salvioli et al. 2009). Second, a history of infectious or autoimmune disease (and associated chronic immune stimulation) is a significant risk factor for the development of acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) (Kristinsson et al. 2011). Third, the extent of hematopoietic decline varies widely between aged individuals, which may partially be attributed to lifestyle choice. Indeed, smoking (Malfertheiner and Schutte 2006) and obesity (Roberts et al. 2010; Renehan et al. 2008) are both linked to higher cancer incidence, while caloric restriction in mice (Anisimov 2009) and prophylactics in humans (i.e., aspirin) (Thakkar et al. 2013; Carlson et al. 2013) have been shown to have antitumor effects. A common feature of these cancer-promoting and cancer-reducing contexts is the observation that they all modulate inflammation, with smoking and obesity promoting inflammation and caloric restriction and aspirin reducing inflammation.
13.3 Aging-Associated Hematopoietic Malignancies in Humans: Forest or the Trees?
Two categories of hematopoietic cancers that show striking increases in incidence in individuals over 65 years of age are leukemias and lymphomas (Fig. 13.3). In the following section we will review three aging-associated leukemias, each thought to initiate within HSCs: chronic lymphocytic leukemia (CLL), chronic myelogenous leukemia (CML), and acute myelogenous leukemia (AML). We will focus on the etiologies of these malignancies and how aging-associated hematopoietic changes in stem and progenitor cell populations may promote disease evolution in older populations.
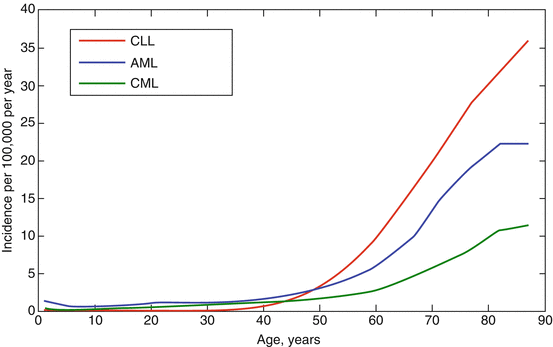
Fig. 13.3
Leukemia incidence in aging populations. The incidence of chronic lymphocytic leukemia (CLL), acute myelogenous leukemia (AML), and chronic myelogenous leukemia (CML) increases drastically after the age of 50 with CLL being the most common leukemia in aged populations (Data are from http://seer.cancer.gov/, for years 1992–2000, and include all sexes and races)
13.3.1 Etiologies of Aging-Associated Cancers
CLL is the most common type of leukemia/lymphoma in adults, with 72 being the median age of diagnosis (Gaidano et al. 2012). CLL is characterized by the accumulation of antigen-stimulated mature B lymphocytes and proliferating precursor cells (Chiorazzi et al. 2005). Although chromosomal translocations and cytogenetic lesions are rare in leukemic clones at earlier points of disease progression, these do appear as the disease progresses (Gaidano et al. 2012; Chiorazzi et al. 2005). Indeed, deletions such as 13q14, 11q22-23, 17p13, 14q32, and 6p21 are found in over half of CLL reported cases (Gaidano et al. 2012). These genetic changes likely have profound biological effects given that they modulate the expression of microRNAs (13q14), p53 (17p13), and the ataxia telangiectasia mutated (ATM; 11q22-23) gene (Chiorazzi et al. 2005). In addition to genetic aberrations in malignant cells isolated from CLL patients, tumorigenic B cells also exhibit altered intracellular signaling. Clinically, CD38 (which augments BCR signaling) or ZAP-70-positive B cells are associated with more aggressive forms of the disease, while decreased activation of these molecules usually indicates a more indolent disease status (Chiorazzi et al. 2005). Recently it has been demonstrated that HSCs from CLL patients are aberrant and contribute to disease progression, which conflicts with the prevailing view that this leukemia initiates in more mature B cells (Alizadeh and Majeti 2011; Kikushige et al. 2011; Damm et al. 2014). In fact, HSCs from CLL patients exhibit cell-intrinsic lymphoid lineage bias that generates clonal CLL-like B cells (Alizadeh and Majeti 2011; Kikushige et al. 2011). Given the clonal nature of B cells that arise from HSCs from CLL patients, it is likely that a selection for oncogenically initiated HSCs occurs, predisposing patients to this malignancy resulting from additional oncogenic events at more B-lineage-committed stages. However, little is known regarding either the initiating genetic/epigenetic events or the aging-associated contexts that favor initiation and progression of this disease.
CML is a clonal disorder of HSCs that results in a malignant increase of myeloid cells, erythroid cells, and platelets in the bone marrow and the peripheral blood. The median age of presentation of CML is 53 years of age (Sawyers 1999). There is no clear genetic or hereditary predisposition for this disease (Garcia-Manero et al. 2003). The acquisition of the Philadelphia chromosome, resulting from a translocation and fusion of the breakpoint cluster region (BCR) gene on chromosome 22 with the ABL gene produces BCR–ABL, a constitutively active, pleiotropic tyrosine kinase capable of promoting cellular growth, proliferation, and survival independent of growth factor regulation (Sawyers 1999). Notably, CML in chronic phase is thought to be a simple leukemia (a myeloproliferative disorder), with Bcr–Abl as the only recurrent mutation (Mullighan et al. 2008). In contrast, the more advanced CMLs and BCR–ABL+ ALLs show numerous additional genetic changes beyond the BCR–ABL translocation (Mullighan et al. 2008), typical of most hematologic malignancies and other cancers. Although the presence of BCR–ABL transcripts is relatively common in the general population, the clinical manifestation of CML only occurs in 1–2 in 100,000 individuals per year (Biernaux et al. 1995; Bose et al. 1998; Matioli 2002; Garcia-Manero et al. 2003). These observations indicate that the acquisition of the driver mutation alone is insufficient to cause disease; thus, additional factors must contribute to disease progression (or lack thereof).
AML is a common form of leukemia in adults with the median age of diagnosis being 69 (Lowenberg et al. 1999). AML refers to a heterogeneous subset of leukemia characterized by clonal expansion of myeloid progenitors (granulocytic, monocytic, erythroid, or megakaryocytic) with a reduced capacity to differentiate into mature myeloid cells. Unlike CML in chronic phase, AML is recognized as a heterogeneous disease with a vast array of driver and secondary mutations required for the development of the disease. These mutations can be broadly classified into two categories: mutations that promote proliferation and those that impair differentiation (Kelly and Gilliland 2002). For the former, activating mutations in FLT3 are the most frequently observed molecular defect in AML, occurring in approximately 30–35 % of adults and 20–25 % of children with AML (Armstrong et al. 2004; Small 2006). Mutations that reduce differentiation include those in genes encoding tet methylcytosine dioxygenase 2 (TET2) and DNA (cytosine-5)-methyltransferase 3 alpha (DNMT3A), which substantially impact on the epigenetic profiles of these cells (Sanders and Valk 2013).
13.3.2 Why Are These Leukemias Associated with Advanced Age?
The field of cancer research is dominated by the view that oncogenesis is rate limited by the incidence of oncogenic mutations and that these mutations are typically advantageous when they occur in the right cell type. Such oncogenic mutations (including activation of proto-oncogenes and deactivation of tumor suppressor genes, whether by genetic or by epigenetic mechanisms) are thought to provide cells with various “Hallmarks of Cancer,” including sustained proliferative signaling and resistance to growth-suppressive and cell death signals (Hanahan and Weinberg 2011). In particular, it is widely accepted that the exponential increase of cancer incidence with age reflects the time required for cells to accumulate sufficient numbers of genetic and epigenetic mutations to confer the cancer phenotype (Kennedy et al. 2012; Vogelstein and Kinzler 2004; Weinberg 2007; Serrano and Blasco 2007; Hoeijmakers 2009; Peto et al. 1975; Nowell 1976). The logic is quite simple: aging leads to mutations (including oncogenic mutations) eventually resulting in cancers such as CLL, CML, and AML. Oncogenic mutations are clearly required for cancer evolution, and increases in genetic/epigenetic diversity in somatic cells associated with aging should contribute to cancer incidence. Increased rates of genomic instability in some cancers can also help promote tumor evolution (Crawford et al. 2004; Agrawal et al. 2012; Chang et al. 2001). However, is the axiomatic attribution of the rising incidence of cancers with age primarily due to the accumulation of oncogenic mutations sufficiently justified? Could it be so simple that the aging-associated increase in cancer incidence is solely explained by the requirement of the time it takes to increase the accumulation of oncogenic mutations?
As referred to above, many, if not most, mutations appear to accumulate during ontogeny, rather than during adulthood (Lynch 1988




Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
