, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
With special contribution from Andrea L. Haws, MD, The Children’s Hospital of San Antonio.
22.1 Langerhans Cell Histiocytosis
22.1.1 Clinical Features
Langerhans cell histiocytosis (LCH) can affect individuals at any age, but the peak incidence occurs at 1–4 years of age [1]. The most classic presentation of LCH is a seborrheic dermatitis-like eruption of the skin. Pink scaly papules and plaques, often with crusting and petechiae, tend to favor the scalp, postauricular area, axillae, and perineum. Less commonly, nodules and ulcerative lesions occur (Fig. 22.1). Mucosal involvement is seen, especially with more severe disease. Erythema and erosions of the gingiva, as well as natal teeth, can be seen. Bony lesions in LCH tend to present with painful swelling. The skull and long bones of the extremities are the most commonly affected sites [2].
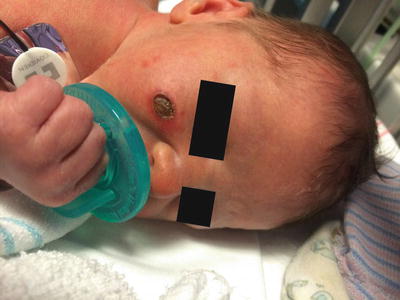
Fig. 22.1
Langerhans cell histiocytosis is characterized by an erythematous, indurated plaque with central ulceration and overlying eschar
In general, LCH involving a single organ has an excellent prognosis and a high rate of spontaneous remission. Up to 50 % of skin LCH lesions regress within a few months [1]. Patients with multisystem or disseminated disease have a much poorer prognosis.
Congenital self-healing reticulohistiocytosis is a rare form of Langerhans cell histiocytosis that presents at birth or during the neonatal period. Cutaneous lesions consist of single or multiple reddish-brown crusted papules or nodules (Fig. 22.2). Although the skin lesions tend to heal spontaneously within days, some patients may have involvement of internal organs, particularly the bones, liver, and spleen [3]. Prognosis depends on the extent of disease.
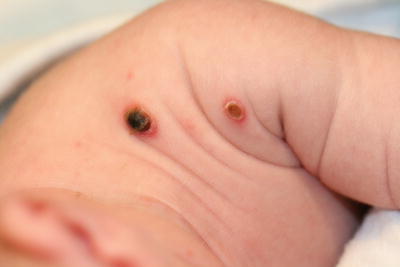
Fig. 22.2
Congenital self-healing reticulohistiocytosis presents as multiple firm, erythematous and ulcerated papulonodules on the shoulder of an infant
22.1.2 Histology
There are many variants of Langerhans cell histiocytoses [4]. While there is great variation in the clinical presentation and clinical course of these subtypes, the histologic changes are largely the same and will be described collectively, concentrating solely on mucocutaneous findings. It should also be noted that involvement of the bone, lung, and pituitary glands might occur in some variants of this process.
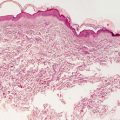
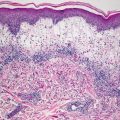
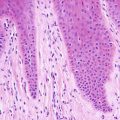
In LCH, the epidermis demonstrates focal parakeratosis overlying varying degrees of spongiosis that is diffuse throughout the epidermis. In most cases, there is abundant exocytosis of Langerhans cells into the epidermis (Fig. 22.3). Langerhans cells can usually be identified based upon cytologic features that include reniform-shaped, grooved, or folded nuclei and abundant cytoplasm (Figs. 22.4 and 22.5). Anti-CD1 a immunohistochemistry is useful in cases when the nature of the infiltrating cells is not readily apparent. Intraepidermal eosinophils are also quite common. Cytologic atypia and significant mitotic activity are rarely encountered. Some cases fail to demonstrate exocytosis of Langerhans cells into the dermis. Treatment with topical corticosteroids prior to biopsy may also suppress exocytosis and mask the diagnostic features.
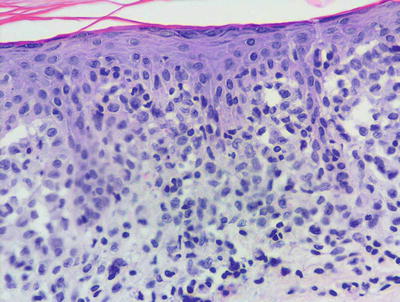
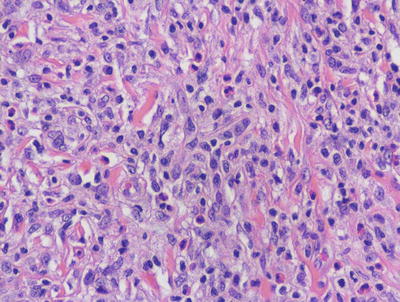
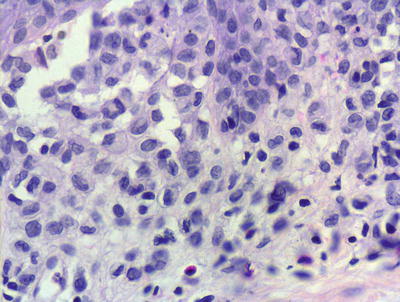
Fig. 22.3
Marked epidermotropism of Langerhans cells into the epidermis is seen in most cases of Langerhans cell histiocytosis
Fig. 22.4
Langerhans cell histiocytosis is comprised of sheets of Langerhans cells with grooved or reniform nuclei and ample eosinophilic cytoplasm. Eosinophils are usually present throughout the infiltrate
Fig. 22.5
Langerhans cells have a characteristic reniform nuclei, often with a grooved appearance. They are readily identifiable on routine H&E sections in most cases, although immunostains can be used to confirm the histologic impression
Within the dermis, there is an infiltrate that consists of Langerhans cells, eosinophils, lymphocytes, and neutrophils. The infiltrate may be diffuse and virtually always involves the papillary dermis. In some cases, extension into the deeper reticular dermis may be present. Less commonly, the infiltrate can appear granulomatous. In these cases, multinucleated giant cells may be present and obscure the true nature of the Langerhans cell process. Touton-type and osteoclast-like giant cells have been described. These same histologic changes have been identified on mucosal surfaces and in the nails when these sites are similarly affected in LCH [5, 6].
Congenital self-healing reticulohistiocytosis demonstrates identical histologic features as described above, and cannot be distinguished from the more progressive LCH by histologic or immunologic means. Clinical observation is the best way to separate these entities. A single case of congenital self-healing reticulohistiocytosis appearing as a solitary nodule has been reported [7]. Rare cases of aggressive congenital Langerhans cell histiocytosis have also been reported. In one such case, the expression of CD56 by the infiltrating cells was noted [8]. Further confounding this distinction, late-onset “congenital” self-healing reticulohistiocytosis has been described as initially appearing in a child 8 years of age [9].
Indeterminate cell disorder demonstrates identical histologic changes to those seen in LCH. However, electron microscopy reveals that Langerhans cells do not contain Birbeck granules, although they share the immunostaining characteristics of traditional Langerhans cells. Rare cases with mucosal involvement have been described [10]. The existence of this variant as a separate and discrete entity has been questioned [11].
Langerhans cell hyperplasia can be seen in many conditions, including some as common as contact dermatitis, and these causes must be excluded before establishing a diagnosis of LCH [12]. Persistent scabetic nodules may also contain abundant Langerhans cells or indeterminate cells [13].
Immunostains can usually distinguish LCH from xanthoma disseminatum, in which the infiltrating cells do not express CD1a [14]. Mast cell disease as it occurs in children may also present some diagnostic dilemma, but there is usually minimal infiltration of the epidermis by mast cells. Giemsa, mast cell tryptase and CD117 stains can also characterize the infiltrating mast cells and differentiate them from Langerhans cells .
22.1.3 Pathogenesis
The pathogenesis of LCH remains enigmatic. A debate persists on whether LCH results from immune dysregulation or malignant transformation of Langerhans cell precursors. LCH has a prominent inflammatory component and can have a benign course, suggesting that LCH may not be a neoplasm. However, supporting evidence that LCH is a neoplasm includes clonality, the presence of the activating BRAF V600E point mutation in addition to KRAS and TP53 mutations, and avoidance of cell death through overexpression of BCL21 and BAX [15, 16].
22.2 Xanthoma Disseminatum
22.2.1 Clinical Features
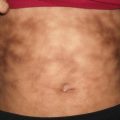
Xanthoma disseminatum is a rare non-Langerhans cell disorder. It may appear at any age and is more common in males than females by 2:1. Three clinical variants have been described: a persistent form, a progressive form with systemic involvement, and a self-limited form.
Cutaneous lesions consist of multiple small orange papules on the face, flexures, and mucosal membranes (Fig. 22.6). The disease course of xanthoma disseminatum is typically benign. Localized lesions may be amenable to surgical excision. Chemotherapy has been used for systemic disease with variable success [17].
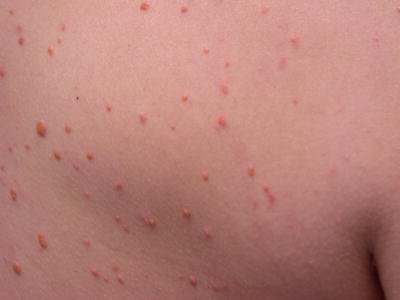
Fig. 22.6
Xanthoma disseminatum is characterized by multiple firm, yellow papules along the trunk
22.2.2 Histology
Histologic changes are those of a dense, confluent infiltrate of histiocytes throughout the papillary and reticular dermis (Fig. 22.7). Mononuclear and multinucleated giant cells with varying amounts of cytoplasmic lipids are admixed with eosinophils and scattered lymphocytes. Touton giant cells are often abundant (Fig. 22.8). Nuclei develop a scalloped appearance due to the presence of cytoplasmic lipid droplets [18, 19]. In very early lesions, lipid may be relatively inconspicuous. In later lesions, fibrosis may be present.
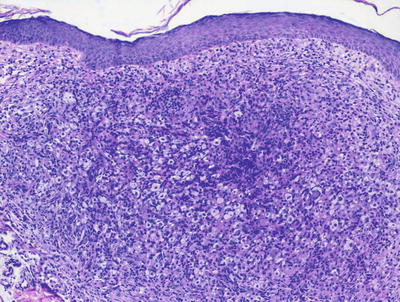
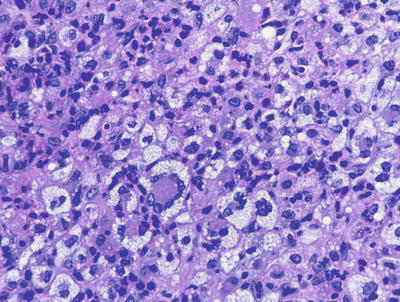
Fig. 22.7
A flattened epidermis overlies a dermis that is distended with sheets of lipid-laden histiocytes admixed with inflammatory cells, including eosinophils in xanthoma disseminatum
Fig. 22.8
Touton-type giant cells are abundant in xanthoma disseminatum
The histologic differential diagnosis of xanthoma disseminatum mainly includes juvenile xanthogranuloma. This entity is histologically indistinguishable, but can easily be separated based upon clinical presentation. A rare entity described as papular xanthoma may also have similar histologic features, but it is easily distinguished based upon clinical presentation [20].
22.2.3 Pathogenesis
Xanthoma disseminatum is a rare disease and belongs to the group of non-Langerhans cell histiocytoses, the pathogenesis for which is unknown. The lipid profiles in these patients are normal [21]. Involvement of the pituitary gland with accompanying diabetes insipidus is present in 30–50 % of patients [21, 22].
22.3 Juvenile Xanthogranuloma
22.3.1 Clinical Features
Juvenile xanthogranuloma (JXG) is a form of non-Langerhans histiocytosis that typically affects infants and young children, but it is occasionally seen in adults. Most lesions are present at birth (20 % of cases) or appear within the first few months of life [2].
A typical JXG lesion is a firm, round papule or nodule, often with a distinctly yellow color (Fig. 22.9). Lesions occur most commonly on the head, neck, and trunk but may be seen on the mucous membranes as well. Lesions are usually solitary (90 % of cases) and asymptomatic. Extracutaneous involvement is possible, with the eye being the most common non-cutaneous site. Other organs are rarely affected [2]. Lesions often spontaneously improve and regress over a period of 3–6 years [23].
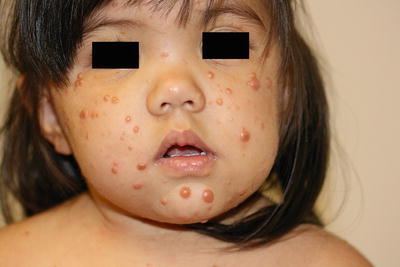
Fig. 22.9
Juvenile xanthogranuloma may present as multiple yellowish-orange papules and papulonodules scattered on the face of a young girl
22.3.2 Histology
Juvenile xanthogranuloma is characterized by a nodular infiltrate of histiocytes within the dermis [24, 25]. The overlying epidermis is either unremarkable or demonstrates slight compression of the rete ridges by the dermal infiltrate in fully developed lesions. Within the dermis, abundant mononuclear and multinucleated histiocytes are present, admixed with an inflammatory infiltrate that contains lymphocytes and variable numbers of eosinophils (Fig. 22.10). The earliest lesions may be difficult to diagnose based upon the lack of visible lipids within histiocytes [26, 27]. In these cases, an infiltrate of mononuclear histiocytes with abundant cytoplasm admixed with scattered eosinophils may be the only findings. As the lesions evolve, abundant lipid is present within histiocytic cytoplasm, resulting in scalloped nuclei and ultimately the formation of Touton giant cells (Fig. 22.11). An inflammatory infiltrate persists in some cases. Late lesions may have dermal fibrosis, and a slightly less pronounced inflammatory infiltrate [28–31].
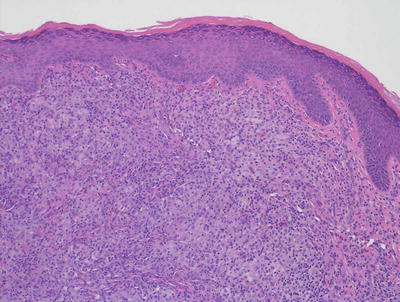
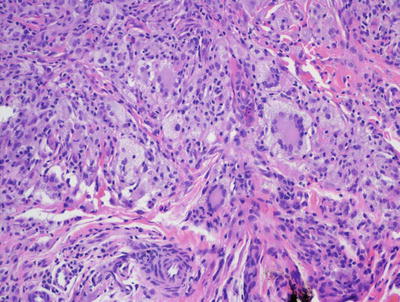
Fig. 22.10
The dermis is filled with histiocytes admixed with inflammatory cells, often including many eosinophils in juvenile xanthogranuloma
Fig. 22.11
Touton giant cells containing abundant peripherally-located cytoplasmic lipid is characteristic of well-formed juvenile xanthogranulomas
Reticulohistiocytoma differs from JXG in lacking significant lipid in histiocytes and the relative paucity of multinucleated cells. Xanthogranuloma disseminatum may be histologically indistinguishable from JXG, but it is easily separated based upon the clinical presentation [32]. Langerhans cell histiocytoses may demonstrate some overlap, but generally display more infiltration of the epidermis by the cellular process. Furthermore, the reniform morphology of Langerhans cells, and the immunostaining profile can easily make this distinction. Other non-Langerhans cell histiocytic proliferations are best distinguished based upon clinical findings [33].
22.3.3 Pathogenesis
Juvenile xanthogranuloma is thought to be a reactive response to a yet unidentified stimulus. Associations of JXG with cafe au lait spots, epilepsy, neurofibromatosis, and childhood leukemias (most commonly juvenile chronic myelogenous leukemia) have been reported, and implicate Ras pathway hyperactivity in this disorder [16, 24]. A recent review highlights the observed association of xanthogranulomas with hematologic malignancies, and suggests that xanthogranulomas are linked to underlying hematologic malignancies through induction of cytokines [34].
22.4 Reticulohistiocytoma
22.4.1 Clinical Features
Reticulohistiocytomas when present as single lesions are clinically indistinguishable from juvenile xanthogranulomas. Reticulohistiocytomas present as papules or nodules, occasionally with a yellow hue.
Multicentric reticulohistiocytosis is rare in children [35, 36]. There may be multiple such lesions that are commonly found overlying joints [37]. In these cases, joint symptoms and mutilating arthritis can occur. While there is a strong association with internal malignancies in adults with this condition, a similar association has not been found in affected children with reticulohistiocytomas. The associated arthritis may respond to treatment with methotrexate, but the cutaneous lesions do not [36].
22.4.2 Histology
Reticulohistiocytoma demonstrates a nodular proliferation of epithelioid histiocytes within the superficial and mid-dermis [38, 39] (Fig. 22.12). This has given rise to the alternate name of epithelioid histiocytoma. The epidermis is either unremarkable or slightly compressed by the dermal infiltrate. Ulceration is distinctly uncommon. The infiltrate consists predominantly of mononuclear histiocytes with abundant granular and eosinophilic cytoplasm (Fig. 22.13). Other cells may contain varying amounts of cytoplasmic lipid. Multinucleated cells, including Touton giant cells, may be encountered, but they are less abundant than in xanthogranulomas. A smaller population of spindle-shaped histiocytes is often seen within the infiltrate. Mitoses are seen in small numbers, but no atypical forms are expected. Admixed lymphocytes, neutrophils, and scattered eosinophils are present. Older lesions have interspersed fibrosis.
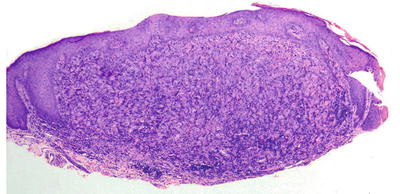
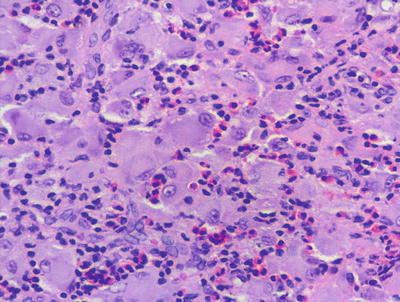
Fig. 22.12
The dermis is filled with an aggregation of large pale-staining histiocytes admixed with inflammatory cells in reticulohistiocytoma
Fig. 22.13
Reticulohistiocytoma is characterized by large mononuclear histiocytes with oncocytic, focally granular cytoplasm and associated inflammatory infiltrate, often with abundant eosinophils. Cytoplasmic lipid is not as prevalent as in xanthogranuloma
The differential diagnosis includes xanthogranuloma, and the lesions may represent a spectrum of a single entity. Cells in xanthogranulomas generally lack the granular eosinophilic cytoplasm, and display more intracytoplasmic lipid. Similarly, xanthogranulomas demonstrate many more Touton giant cells than are usually encountered in reticulohistiocytoma. The differential diagnosis is unimportant in most cases, and only becomes significant if a diagnosis of multicentric reticulohistiocytosis is being considered in association with internal malignancy. However, this entity is very rare in children [35]. A Spitz nevus can also enter into the differential diagnosis, but it can be discriminated from reticulohistiocytoma based upon the nesting pattern, the intraepidermal component, and the progressive maturation of cells with descent into the dermis. S100 or melanocyte marker immunostain can also resolve the differential diagnosis if needed.
22.4.3 Pathogenesis
Reticulohistiocytoma is thought to be a disorder of macrophage cells involving non-Langerhans histiocytes, which are stimulated by some yet unknown precipitating factor [38]. Although a neoplastic process has not been excluded, it likely represents a reactive process, resulting in a localized inflammatory reaction. Various precipitating factors have been proposed, including infections, inflammation, as well as autoimmune and paraneoplastic processes [40]. The presence of a large number of neutrophils in some lesions suggests an infectious etiology, although an infectious agent has not yet been identified in these lesions [38].
22.5 Benign Cephalic Histiocytosis
22.5.1 Clinical Features
Benign cephalic histiocytosis is a form of non-Langerhans cell histiocytosis that is seen primarily in infants and young children. The average age of onset is 15 months and approximately 45 % of cases occur in infants less than 6 months old [2].
Typical lesions are small (up to a few mm) yellow-brown macules and thin papules that occur mainly on the head, face, and neck (Figs. 22.14 and 22.15). A small percentage of children develop lesions on the trunk and extremities [2]. Although this condition has shown some overlap with other histiocytic disorders, it generally has a benign course. Lesions regress over months to a few years, but may leave flat or atrophic hyperpigmented macules behind [41].
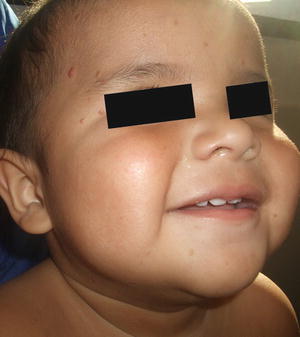
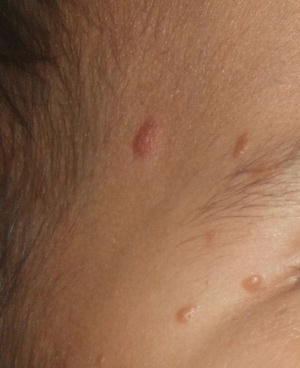
Fig. 22.14
Benign cephalic histiocytosis may present as multiple reddish-brown papules scattered on the face in this young boy
Fig. 22.15
Benign cephalic histiocytosis , a closer view of lesions in the same patient in Fig. 22.14
22.5.2 Histology
Biopsies taken from patients with benign cephalic histiocytosis display a well-circumscribed proliferation of histiocytes centered mainly in the upper half of the dermis with no tendency for upward migration [42]. Mononuclear histiocytes are the predominant cell type with little cytoplasmic lipid. Touton giant cells may be observed, but they are relatively scant in most cases.
Ultrastructural analysis has demonstrated comma-shaped bodies in the cytoplasm of infiltrating histiocytes [41, 43, 44]. The existence of benign cephalic histiocytosis as a distinct entity has been called into question, with some authors suggesting that it represents a variant of generalized histiocytosis or juvenile xanthogranuloma [26, 45, 46]. The differential diagnosis includes other non-Langerhans cell histiocytic proliferations, and the distinction is primarily made based on the clinical presentation and disease course.
22.5.3 Pathogenesis
The etiology of benign cephalic histiocytosis is unclear. It is a diagnosis that seems to belong in the spectrum of non-Langerhans histiocytoses [42]. It has also been suggested that benign cephalic histiocytosis represents a localized form of generalized eruptive histiocytoma [41]. A recent report describes a case of accelerated regression of benign cephalic histiocytosis with antibiotic administration. Interestingly, Staphylococcus aureus was isolated from the dermal histiocytic collections, and the affected child was found to have low plasma levels of IgA [47].
22.6 Progressive Nodular Histiocytosis
22.6.1 Clinical Features
Progressive nodular histiocytosis is more common in adults than in children [48]. Typically, there is a generalized eruption of hundreds of small yellow-brown papules as well as scattered, larger subcutaneous nodules. Lesions on the face may coalesce to give a “leonine” appearance. Larger nodules may develop ulceration and bleeding, which can be painful. There can be mucous membrane involvement [2]. The disease course is typically chronic and progressive, although it is generally benign with patients usually remaining in relatively good health [48].
22.6.2 Histology
Histologic findings of progressive nodular histiocytosis are those of a predominantly spindle-shaped histiocytic infiltrate that is histologically identical to a spindle cell xanthogranuloma [49, 50]. Rare cytoplasmic lipid may be seen, but it is usually quite focal. Systemic involvement with histiocytic tumors has been reported and helps to differentiate this entity from similar limited cutaneous eruptions [51, 52]. Most authors believe progressive nodular histiocytosis to be part of a spectrum of non-Langerhans cell histiocytoses and is closely related to xanthogranulomas [46, 52, 53]. The histologic differential diagnosis includes a dermatofibroma with focal lipidization [53].
22.6.3 Pathogenesis
The pathogenesis of this rare disease is unknown. Cytogenetic analysis of a patient with progressive nodular histiocytosis revealed a normal karyotype [49]. It has been debated whether this is a reactive or neoplastic process .
22.7 Sinus Histiocytosis with Massive Lymphadenopathy
22.7.1 Clinical Features
Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) is a disorder that primarily affects young children [54]. It typically presents with massive lymphadenopathy, which affects the cervical region in up to 90 % of affected individuals [54]. Extranodal disease can occur, and the skin may be affected, typically appearing as nonspecific papules and nodules. Sinus histiocytosis with massive lymphadenopathy is typically a self-limited disease and spontaneously regresses [54].
22.7.2 Histology
The cutaneous lesions in Rosai-Dorfman disease are histologically quite similar to changes described in the more common lymph node-based sinus histiocytosis with massive lymphadenopathy. A dense infiltrate of inflammatory cells fills much of the dermis with no tendency for invasion of the epidermis or cutaneous appendages [55, 56] (Figs. 22.16 and 22.17). Admixed with sheets of histiocytes are lymphocytes, neutrophils, plasma cells, and occasional eosinophils [57]. The histiocytes resemble those seen in lymph node sinuses and demonstrate emperipolesis, in which histiocytes engulf other inflammatory cells [58, 59]. While this finding is not pathognomic for the disease, it is a very characteristic finding in this entity, and is usually readily apparent on H&E staining. Plasma cells are often found rimming blood vessels within the midst of the lesions. Thick-walled vessels and lymphoid nodules have been described at the periphery of lesions [55].
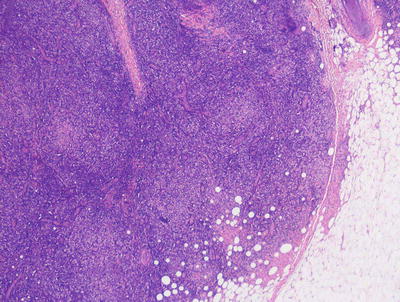
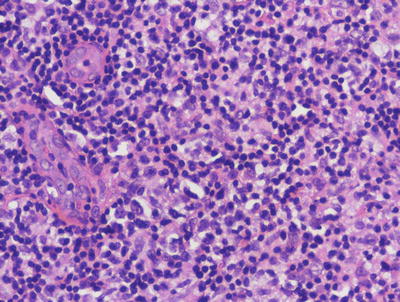
Fig. 22.16
The dermis is filled with large histiocytes admixed with a dense inflammatory infiltrate. The infiltrate may extend into the subcutis in sinus histiocytosis with massive lymphadenopathy
Fig. 22.17
Large histiocytes may demonstrate emperipolesis, and may be surrounded by a dense collection of lymphocytes with occasional plasma cells and eosinophils in sinus histiocytosis with massive lymphadenopathy. Endothelial cells in dermal vessels are edematous and resemble small granulomas
When purely cutaneous, these lesions can be mistaken for metastatic melanoma, a problem further confounded by the diffuse S100 positivity of the histiocytes in this disorder [60]. Careful attention to morphologic detail, clinical history, and other immunostains will allow for distinction. The differential diagnosis also includes infectious granulomatous processes, xanthogranuloma, reticulohistiocytoma, other histiocytic accumulations, and lipidized dermatofibroma [61, 62]. Histiocytes in these other entities do not express S100, and the pattern of plasma cellular cuffing of vessels, and histiocytes with marked emperipolesis would not be the expected findings .
22.7.3 Pathogenesis
The systemic form of sinus histiocytosis with massive lymphadenopathy is thought to involve an abnormal histiocytic response to a viral stimulus (such as Epstein-Barr virus and human herpesvirus 6 or 8) [63]. There has been a report of regression with acyclovir administration [64]. The disorder is associated with abnormalities of the immune system, such as infection, autoimmune disease, leukemia/lymphoma, and HIV [63]. Molecular investigation of a case of generalized pure cutaneous form of Rosai-Dorfman disease indicates that the histiocytes may have undergone a transition to mesenchymal cells, having both macrophage and mesenchymal characteristics, which may best be classified as a histiocyte-mesenchymal transition [65]. The molecular underpinning of sinus histiocytosis with massive lymphadenopathy may be related to the findings of germ line mutations in SLC29A3 , which encodes an intracellular equilibrative nucleoside transporter (hENT3) with affinity for adenosine. Mutations in SLC29A3 have been found in familial histiocytosis syndrome and familial sinus histiocytosis with massive lymphadenopathy [66]. It is not known whether SLC29A3 is involved in sporadic disease .
22.8 Mast Cell Disease
22.8.1 Clinical Features
Fifty-five percent of patients with cutaneous mast cell disease present before 2 years of age, and about 10 % of cases appear between 2 and 15 years of age [67]. Three variants of mastocytosis tend to affect children: solitary mastocytoma, urticaria pigmentosa, and diffuse cutaneous mastocytosis. Solitary lesions are present at birth in approximately 40 % of cases, and typically appear by 1 year of age. Lesions appear as reddish-brown macules, patches, papules, or plaques that have a tendency to urticate with pressure or friction (Fig. 22.18). Urticaria pigmentosa is the most common form of cutaneous mastocytosis in children. It presents with diffuse reddish-brown macules and papules (Fig. 22.19). Diffuse cutaneous mastocytosis is the least common type, characterized by skin that may appear normal to diffusely doughy, reddish brown or have a “peau d’orange” texture [67]. Individual lesions may become irritated and pruritic, occasionally even forming a blister. Patients with more extensive skin lesions or systemic involvement can have gastrointestinal and respiratory symptoms as well as more severe allergic reactions. There is a tendency for individual skin lesions to improve over time. Typically, many lesions involute by puberty [67].

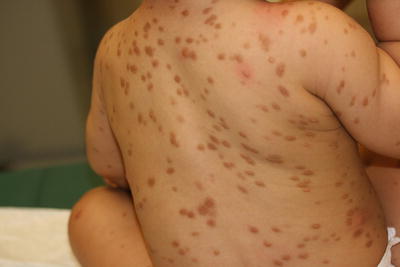
Fig. 22.18
Mastocytoma often presents as an erythematous, faint orange plaque with peau d’orange surface changes
Fig. 22.19
Urticaria pigmentosa presents as scattered brown macules and papules on the torso and extremities of an infant. Some lesions demonstrate localized redness and edema after being stroked (Darier’s sign)
22.8.2 Histology
Mast cell disease has a wide range of clinical patterns. However, the histologic manifestations are quite similar in each of these forms, differing solely in the density of dermal mast cells. A common finding is basilar hyperpigmentation with minimal increase in melanocytes but marked increase in melanin throughout the basal melanocytes [68]. Within the dermis, increased numbers of mast cells may appear as small perivascular clusters of fusiform or cuboidal cells, making it difficult to discern from lymphocytes (Figs. 22.20 and 22.21). Alternatively, large sheets of cuboidal cells may fill the dermis. In most cases of mast cell disease independent of the number of mast cells, there are scattered eosinophils within the infiltrate and varying amounts of dermal edema. Later lesions may also show fibrosis and some necrosis [69]. In children, it is uncommon for mast cells to demonstrate cytologic atypia, pleomorphism, or markedly increased mitotic activity [70, 71].
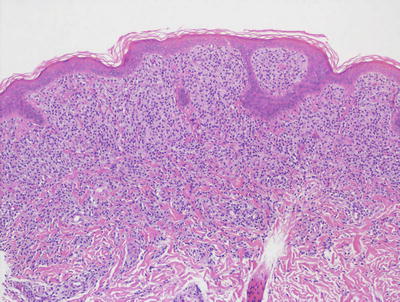
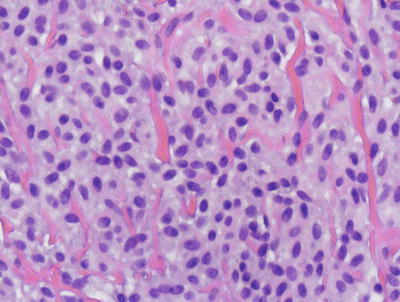
Fig. 22.20
The dermis is filled with monotonous-appearing cuboidal cells with central nuclei and abundant cytoplasm in mastocytosis . In children, a tumoral infiltrate is more common than the variants of the disease seen in adults
Fig. 22.21
Mast cells in mastocytosis appear as monotonous sheets of cuboidal to slightly spindled cells with amphophilic cytoplasm. There is some resemblance to melanocytic nevus cells, although the nesting pattern in a nevus makes the distinction straightforward
There have been many attempts to quantify the number of dermal mast cells required to establish a diagnosis in more subtle cases. These include attempts at absolute cell counts, counts per unit area around dermal blood vessels, and minimum percentages of dermal inflammatory cells [72]. No formulas have yet proven to be completely reproducible. In these cases, it is best to have a biopsy with affected skin adjacent to clinically normal skin, or two adjacent biopsies in order to provide a “normal” control against which to compare mast cell numbers.
The differential diagnosis of mastocytosis varies with the number of dermal mast cells. In cases with a relatively scant infiltrate, the differential diagnosis includes entities that have a superficial and deep perivascular infiltrate with scattered eosinophils and dermal edema, such as an arthropod bite reaction and a drug eruption. If only minimal dermal infiltrate is present, the basilar hyperpigmentation may raise the differential diagnosis of a lentigo simplex. When it is difficult to identify precisely the nature of the dermal infiltrate, special stains such as Giemsa stain and immunostains for c-Kit (CD117) and mast cell tryptase may be helpful in making these distinctions and resolving the etiology of the cells [73].
In cases with sheets of mast cells infiltrating the dermis, the appearance may resemble an intradermal nevus. The lack of dermal pigmentation, maturation, and a nesting pattern should all help to make the distinction fairly straightforward. Other hematopoietic infiltrates may also enter into the differential diagnosis, but use of special stains and other immunomarker studies should serve to readily distinguish these other entities .
22.8.3 Pathogenesis
Mast cells express c-Kit (CD117 ), which is a receptor tyrosine kinase that plays a crucial role in mast cell development. It is a transmembrane receptor for stem cell factor (SCF) . The interaction between c-Kit and SCF seems to play an essential role in the development of mastocytosis [74, 75]. Most mastocytosis cases are associated with somatic gain of function mutations in the c-Kit gene, resulting in constitutive activation of c-Kit as well as augmented mast cell proliferation and survival [74, 76]. The most common gene mutation is D816V, and it is detected in more than 95 % of adult patients with systemic mastocytosis, and in about one-third of pediatric patients with cutaneous mastocytosis [67, 77]. In addition to c-Kit, activating mutations in N-Ras have been identified in patients with systemic disease. Clonal mast cells in some of these patients harbor both N-Ras and c-Kit mutations [78]. These findings suggest that N-Ras mutations may have the potential to precede c-Kit mutations in clonal development. Since mast cell survival is dependent on c-Kit, it has been proposed that the presence of these two activating mutations may have a greater impact on the expansion of mast cells and disease severity [78].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


