Fig. 2.1
Analysis of rearrangements of the T-cell receptor (TCR) beta chain by polymerase chain reaction (PCR). The gene scan on the top shows amplification of segments as multiple small peaks and a large predominant peak. This pattern is consistent with a monoclonal population of T-lymphocytes. In the right context, this supports a neoplastic population of T lymphocytes. The bottom of the Figure shows a polyclonal pattern characterized by multiple small peaks, consistent with the presence of reactive, nonneoplastic T lymphocytes. TCR beta chain analysis is especially useful in the identification of a monoclonal peak in a polyclonal background
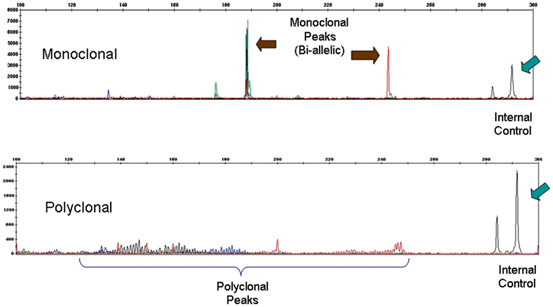
Fig. 2.2
Analysis of rearrangements of the T-cell receptor (TCR) gamma chain by polymerase chain reaction (PCR). In this case the scan on the top shows that the amplification of segments yielded multiple small peaks and two predominant peaks, consistent with a bi-allelic monoclonal population of T-lymphocytes. The bottom shows analysis of TCR gamma chain gene rearrangement in a different specimen. It reveals multiple small peaks, consistent with a polyclonal population of likely reactive, nonneoplastic T lymphocytes. As with the TCR beta chain gene rearrangement analysis, the correct interpretation of this result should be made in conjunction with clinical and pathological findings. The arrows indicate internal controls
The sensitivity of PCR-based tests varies depending on the type of sample and technical issues. It is accepted that the minimum percentage of detectable clonal cells by this method is around 1 % [11] and that its overall sensitivity and specificity is around 70 and 97 % for mycosis fungoides (MF), respectively [12]. False negative results may be due to low numbers of malignant T-cells or absence of TCR gene rearrangement in the lymphoma cells [13]. False positivity (pseudoclonality) may result from amplification of TCR gene rearrangement present in a few T-cells composing a sparse, reactive lymphocytic infiltrate [14]. Duplicate analyses may distinguish reactive from neoplastic proliferations since the dominant peaks detected in reactive conditions vary within the same sample while true clonal peaks are usually reproducible [15].
In recent years, great value has been given to the demonstration of the presence of identical T-cell clones at different anatomical sites as a highly specific tool in discriminating between MF and inflammatory conditions [16], the so-called stable clonal pattern [14]. However, this pattern has also been reported in some inflammatory and “borderline” processes [17]. Conversely, genetically unstable subclones have been described in T-cell lymphomas [18] and clonal heterogeneity in MF lesions from distinct anatomical sites has been reported [19, 20]. The utility of comparing clones from different anatomical sizes for the diagnosis of T-cell lymphoma needs to be evaluated in the context of the clinical and histopathological findings .
Immunoglobulin Gene Rearrangement Analysis
The genes encoding immunoglobulins (Ig), the antigen receptors in B-cells, include the heavy-chain (IGH), kappa light chain, and lambda light chain genes. The IGH gene rearrangement starts with the combination of a diversity (DH) segment with a joining (JH) segment, which then is joined with a variable (VH) segment to create a VDJH sequence. The kappa gene rearrangement then follows. Depending on the functionality of the rearranged kappa alleles, the lambda gene will be rearranged, usually after deletion of both kappa genes [21].
PCR-based methods for detection of IGH gene rearrangement utilize four sets of consensus primers designed to amplify three conserved framework regions within VH and one within JH. From these, PCR amplification of framework region III in the VH and the framework region in JH segments is the most widely used test in clinical practice, due to the low molecular weight of the resulting amplicon and therefore the possibility of using formalin-fixed paraffin-embedded tissue as DNA source [22].
Same as for TCR gene rearrangement analysis, the peaks obtained by capillary electrophoresis are evaluated for the presence of a dominant population or populations (Figs. 2.3 and 2.4). The sensitivity of the method, although usually high, varies according to a number of factors such as the number of reactive B-cells present in the background and tissue fixation, and it can be as low as 47 % in formalin-fixed paraffin-embedded tissue [23]. The rate of false negative results seems to be especially high in cases of diffuse large B-cell lymphoma (DLBL) and follicular lymphoma, which have a high frequency of somatic mutations [24]. These mutations lead to sequences that are noncomplementary to the primer sequences. Such false-negative result is avoided by using multiple PCR target regions such as FR1, FR2, and FR3 segments of IGH gene. Another way to increase the sensitivity to detect clonal B-cell populations is by adding a test for immunoglobulin light chain (IGL) kappa or lambda gene rearrangement [25].
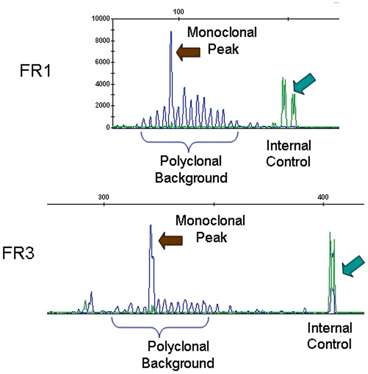
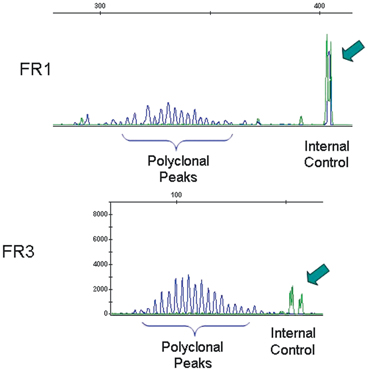
Fig. 2.3
Analysis of rearrangements of the immunoglobulin heavy chain (IGH) genes by polymerase chain reaction (PCR). Evidence of rearrangements of variable, diversity, and joining (VDJ) regions of IGH can be visualized by using pairs of primers at the V and J regions. Each lymphocyte in the analyzed specimen has a rearrangement that renders a segment that is amplified. In B-cell neoplastic processes, the presence of a prominent or distinct peak reflects that a significant number of cells in the specimen have the same size of amplified product. This finding constitutes a monoclonal peak and it usually correlates with a neoplastic expansion of B-cells. The use of three primers for the framework region (FR) of the V regions yields a higher chance of identifying a monoclonal population. The top of the figure shows the results of PCR amplification using FR1 and J region primers, while the bottom shows the amplification using FR3 and J region primers. Both analyses yielded the identification of a monoclonal peak amidst several smaller peaks, indicating that the neoplastic clone is admixed with reactive B lymphocytes. The size of the amplified segment is 98 base pairs on the top and is 320 base pairs on the bottom. Internal controls are used to confirm the size of segments and that amplification is taking place. The arrows indicate internal controls
Fig. 2.4
Analysis of rearrangements of the immunoglobulin heavy chain (IGH) genes by polymerase chain reaction (PCR). In a reactive process, most lymphocytes render different size of amplified product and appear as multiple peaks, interpreted as polyclonal peaks/polyclonal background. In this case, the amplification of segments using FR1 and FR3 primers revealed a polyclonal pattern, consistent with a reactive, not neoplastic infiltrate. Similar results were obtained using FR2 primers. Correlation with clinical and pathological findings is still needed for an adequate interpretation of results of IGH gene rearrangement analysis. The arrows indicate internal controls.
False positive results may be seen in cases in which the skin biopsy shows only sparse infiltrate and the PCR products amplified from the few B-cells appear as a distinct peak. Again, as for T-cell pseudoclonal cases, repeated testing may help in discriminating false clonality from true clonal B-cell expansion. Cases of cutaneous lymphoid hyperplasia have been demonstrated to be clonal for IGH gene rearrangement [26], and clonal IGH gene rearrangement has been reported in T-cell proliferations [27]. Other causes of false-positive results include immune disorders and infections showing a predominant B-cell population .
It is important to emphasize that the demonstration of monoclonal IGH or TCR gene rearrangement in a cutaneous lymphoid proliferation by itself does not make the diagnosis of lymphoma, and that both positive and negative results need to be interpreted in the appropriate clinical and histopathological context .
Detection of Chromosomal Translocations
From the several chromosomal translocations identified in hematolymphoid proliferations only a few are routinely investigated in the dermatopathology practice.
The t(14:18)(q32;q21) IGH/BCL2 translocation can be analyzed by FISH and PCR. Fluorescence in situ hybridization (FISH), using appropriate probes, can detect almost all BCL2 translocations [28]. This translocation occurs in more than 85 % of cases of follicular lymphoma [29] but in only a small number of primary follicular lymphomas of the skin [30], and therefore its presence should suggest secondary involvement of the skin by systemic follicular lymphoma.
Translocations associated with extranodal marginal zone lymphoma rarely occur in the primary cutaneous tumors. The t(14:18)(q32;q21) MALT1/IGH has been identified in a subset of extranodal marginal zone lymphomas of the skin [31].
The t(2;5)(p23;q35)ALK/NPM, found in most of the cases of systemic anaplastic large cell lymphoma (ALCL) [32], has been rarely reported in primary cutaneous ALCL [33]. The demonstration of ALK gene rearrangements by FISH is thus very suggestive of secondary cutaneous involvement by systemic ALK+ ALCL [34]. A subset of primary cutaneous ALCL harbors (interferon regulatory factory-4) translocations; [35] FISH testing in this setting may be of potential diagnostic utility (Fig. 2.5) .
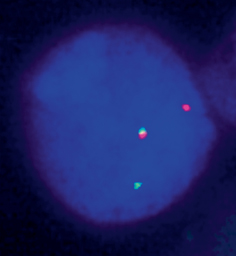
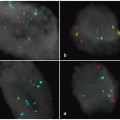
Fig. 2.5
Break-apart fluorescence in situ hybridization (FISH) analysis for identification of translocations involving DUSP22-IRF4 gene locus. The picture shows a nucleus of a cutaneous anaplastic large cell lymphoma (C-ALCL) cell in which there is separation of red and green signals flanking the locus containing the DUSP22 and IRF4 genes, indicating a translocation involving this locus. This finding is most often seen in primary cutaneous ALCL, though it may be seen occasionally in systemic ALK-negative ALCL. (Courtesy of Dr. Andrew L. Feldman, Mayo Clinic, Rochester, MN, USA)
In situ Hybridization Studies
Demonstration of cytoplasmic immunoglobulin (Ig) light chain mRNA in plasma cells can be achieved by in situ hybridization (ISH) studies performed on formalin-fixed paraffin-embedded tissue sections [36]. Restricted Ig lambda or kappa light chains support a diagnosis of extranodal marginal zone lymphoma.
Epstein-Barr virus-encoded mRNA (EBER) ISH studies are important in a number of T-cell and B-cell lymphoproliferative disorders and lymphomas. Latent Epstein-Barr virus (EBV) infection can be revealed by ISH studies due to the large load of EBERs present in the nuclei of infected cells [37].
T-cell Lymphoid Proliferations of the Skin
Mycosis Fungoides
Mycosis fungoides (MF) is the most common cutaneous T-cell lymphoma. Clinical features are crucial for diagnosing this entity, especially on its early stages. The initial presentation is that of small scaly patches, usually on sun-protected areas such as the buttocks, lower abdomen, and thighs. In a minority of patients the patches disseminate and indurated raised plaques appear. The third stage is called tumor stage, which results in nodules that frequently ulcerate and may involve sun-exposed skin. Extracutaneous dissemination may be seen as the disease progresses. The histopathological findings on early, patch-stage mycosis fungoides are subtle and can be easily overlooked. The lymphocytic infiltrate is usually composed of small lymphocytes with minimal or no atypia, arranged around vessels within the papillary dermis and superficial plexus. Epidermotropism by lymphocytes, considered a strong sign for suspecting MF (especially in the absence of important spongiosis), may be very focal. As the lesion progresses, the lymphocytes start to display more obvious cytologic atypia. Plaques of MF usually exhibit cells with “cerebriform” nuclei and often show Pautrier’s microabscesses. Dense infiltrates occupying the reticular dermis characterize tumor lesions. The lymphocytes in tumor stage MF may display an anaplastic morphology making the distinction between MF and ALK negative anaplastic large cell lymphoma (ALCL) impossible without clinical information. Many clinical and histopathological variants of MF have been described. Although scoring systems using clinical, histopathological, immunophenotypic, and molecular characteristics are in use [38], the diagnosis of early MF can still be cumbersome.
Staging of MF, mainly based on clinical and histopathological findings, correlates with prognosis [39]. Immunophenotyping is used as an adjunct for diagnosis. Most of the cases display a CD3+, CD4+, CD8− phenotype. Demonstration of loss of CD2, CD3, CD5, or CD7 may help in the diagnosis [11]. However, some controversy still exists about the exact role of immunohistochemistry in the diagnosis of early MF, since overlapping findings may be seen in inflammatory conditions [40] and the prognosis of early MF does not seem to be influenced by phenotype [41].
Molecular studies can be helpful in the diagnosis of MF, if taken in conjunction with clinical and histopathological findings. T-cell receptor (TCR) gene rearrangement studies are used to demonstrate T-cell clonality in MF [42]. Although Southern blot analysis is still considered the gold standard for determination of T-cell clonality, [43] PCR-based methods are routinely used for this purpose. Investigation of TCR-γ chain gene rearrangements is the most commonly used test in clinical laboratories, due to its high sensitivity [44, 45]. Evaluation of TCR-β gene rearrangements is being increasingly performed in clinical settings. It is expected that the assessment of both TCR-γ and TCR-β gene rearrangements will increase the sensitivity of T-cell clonality detection in MF [8].
When interpreting TCR gene rearrangement studies the dermatopathologist should be aware of a few caveats. Inflammatory conditions in which the lymphocytic infiltrates are scant not uncommonly yield oligoclonal or even monoclonal bands [3]. Duplicate analyses are needed to rule out the possibility of pseudoclonality in these cases [14]. On the other hand, the sensitivity of the PCR-based methods used for TCR gene rearrangement determination widely varies: it is not uncommon that patch MF lesions show oligoclonality [14]. Therefore, a negative or oligoclonal TCR gene rearrangement pattern does not rule out MF.
Sézary Syndrome
Sézary syndrome (SS) is characterized by erythroderma, lymphadenopathy, and the presence of the so-called Sézary cells in peripheral blood. Controversy still exists about the distinction between erythrodermic MF (a condition that may evolve through patch/plaque disease and has variable levels of blood involvement) and SS [46]. Some evidence suggests that these are two different processes and that they constitute malignancies of thymic memory T cells (SS) and skin resident effector memory T cells (MF) [47]. The prevailing view of SS as the leukemic counterpart of MF seems therefore not completely accurate. On clinical grounds, however, there is frequent overlapping between these two entities and a diagnosis of SS should be made under strict criteria, such as the demonstration of a T-cell clone in peripheral blood, peripheral blood lymphocytosis with a CD4+/CD8+ ratio greater than 10, and circulating Sézary cells greater than 1 × 109/L.
Histologically, the changes seen in SS may be indistinguishable from those seen in MF. However, lack of epidermotropism and low grade cytologic atypia are common in SS [48]. Most of the cases exhibit a CD3+, CD4+, CD8− phenotype.
Clonal TCR gene rearrangements are commonly demonstrated in peripheral blood of patients with SS [1]. These monoclonal TCR gene rearrangements can be identical to those detected in skin [49] and this finding has been used as evidence of multiple site involvement by the same malignant process. However, different clonal TCR gene rearrangements in peripheral blood and skin have been detected in a number of patients, suggesting clonal heterogeneity in some cases [14, 19]. Moreover, monoclonal T-cell populations in peripheral blood have been associated with autoimmune disorders [17], advanced age [50], and can be identified in association with other non cutaneous lymphomas and inflammatory skin conditions [1]. Patients with SS may be negative for T-cell clones in peripheral blood [51].
Primary Cutaneous CD30-Positive T-cell Lymphoproliferative Disorders
Primary cutaneous CD30-positive T-cell lymphoproliferative disorders (CD30+ TLPD) constitute the second most common cutaneous T-cell lymphoma/lymphoid proliferations. The WHO classification recognizes three types: primary cutaneous anaplastic large cell lymphoma (C-ALCL), lymphomatoid papulosis (LyP), and borderline lesions. It is recognized that these disorders represent a clinical and histological spectrum of entities going from LyP, a recurrent, benign diffuse eruption of papulonodular lesions that regress spontaneously, to C-ALCL, usually presenting as one or multiple localized ulcerated nodules they may in some cases regress [52]. Distinction should be made from systemic ALCL with secondary involvement of the skin and from other lymphomas showing CD30 expression, such as mycosis fungoides with large cell transformation [53].
LyP has been classified classically in three histological types [54]. Type A, that resembles Hodgkin lymphoma, i. e., shows aggregates of large Reed-Sternberg-like CD30-positive cells in a background of inflammatory cells; type B, or papular mycosis fungoides-like; and type C, or ALCL-like in which the large CD30-positive cells are arranged in sheets with minimal inflammatory background. Recently, a “type D” denomination has been proposed for CD8-positive lesions [55]. Cutaneous anaplastic large cell lymphoma (C-ALCL) reveals a dermal infiltrate composed of large anaplastic, pleomorphic cells, most of them expressing CD30.
The demonstration of clonal rearrangements of TCR in CD30+ TLPD has been variably reported [56, 57]. It is important to take in consideration that identical clones can be detected in different CD30+ TLPD lesions [58] as well as in LyP and concomitant lesions of mycosis fungoides [57]. When dealing with ALCL lesions, it is important to distinguish C-ALCL from involvement of skin by systemic ALCL. Cutaneous anaplastic large cell lymphoma (C-ALCL) cases usually lack t(2;5) and ALK expression by immunohistochemistry [59]. Translocations involving IRF4 detected in C-ALCL but not in systemic ALCL, have been reported as useful in this differential diagnosis [35].
Other Primary Cutaneous T-Cell and NK Lymphomas
This group encompasses rare lymphomas in which a diagnosis of MF has been excluded, clinically and histopathologically .
Primary cutaneous pleomorphic CD4-positive small/medium sized T-cell lymphoma is a provisional category in the 2008 WHO classification. There is controversy about whether this entity should be considered a true lymphoma or an atypical lymphoid proliferation of undetermined significance [60]. It presents as a single or multiple localized lesions on the upper trunk, neck, or face. The course is usually indolent although some aggressive cases have been reported [61]. Histologically, a dermal infiltrate composed of small to medium-sized T cells expressing CD4 admixed with numerous B cells, variable number of CD8-positive T cells, plasma cells, eosinophils, and histiocytes, is present. Expression of BCL6, PD-1, and CXCL13 has been reported in these cases and linked this lesion to a possible follicular T helper origin [62]. In a large series of cases, 60 % of cases were found to have a monoclonal rearrangement of the TCR gamma [60].
Primary cutaneous CD8-positive aggressive epidermotropic cytotoxic T-cell lymphoma (Berti’s lymphoma) on the other hand, is characterized by multiple skin nodules that rapidly disseminate [63]. The prognosis is usually poor. On histological examination, the tumor cells—which can range from small to large—show marked epidermotropism and cytotoxic phenotype. Monoclonal TCR gene rearrangement has been reported in most of the few reported cases [64, 65].
Subcutaneous panniculitis-like T-cell lymphomas (SPTCL) include only cases with the alpha-beta phenotype [66]. Patients usually present with nodules or plaques on lower extremities with or without systemic symptoms. A common association with hemophagocytic syndrome was originally reported [67]. The skin biopsies show a lobular panniculitis pattern. Epidermal or marked dermal involvement is uncommon. The infiltrating cells are an admixture of small, medium, or large cells with variable numbers of histiocytes. Rimming of adipocyte spaces by lymphoma cells, erythrophagocytosis, fat necrosis, and karyorrhexis is variably seen. The atypical cells express CD3 and CD8 and are usually negative for CD4. Distinction of this entity from inflammatory conditions such as lupus erythematosus profundus, Weber-Christian disease, histiocytic cytophagic panniculitis, and reactive panniculitis to drugs or injections can be very challenging. Molecular analysis of TCR gene rearrangement helps in the differential diagnosis, since most of the cases of SPTCL are monoclonal [68].
Primary cutaneous gamma-delta T-cell lymphoma is an aggressive tumor that occurs in young adults and presents with multiple lesions, commonly on proximal lower extremities, and shows frequent mucosal involvement [69]. Three histologic patterns have been described: epidermotropic, dermal, and subcutaneous [70, 71]. These patterns may be observed within a single biopsy. Epidermotropism may be present. Rimming of adipocytes, similar to that seen in SPTCL, may be seen [70]. The tumor cells have a cytotoxic phenotype, with strong expression of granzyme B, TIA-1, and perforin. The T-cell phenotype of the tumor cells is confirmed by expression of CD3. The cells are usually double negative for CD4 and CD8, [72] although CD8 expression has been observed in some cases. CD56 is commonly positive. The tumor cells are negative for βF1 and positive for TCRγ and TCRδ by immunohistochemistry (the latter requires frozen tissue.) Most of the cases show monoclonal TCR gene rearrangement [71].
Cutaneous adult T-cell leukemia/lymphoma (ATLL) refers to skin-limited lesions without lymph node or peripheral blood involvement [73, 74]. ATLL is endemic in Japan, the Caribbean islands, South America, and regions of Central Africa. It is caused by the human T-cell leukemia virus type I (HTLV-1) [73]. Secondary cutaneous involvement is frequently seen in the systemic form. On histologic examination, ATLL cells in skin display a predominantly perivascular distribution and varying degrees of epidermotropism in a pattern sometimes indistinguishable from that seen in mycosis fungoides. The tumor cells are positive for CD3, CD4, and CD25 [75]. Monoclonal integration of HTLV-1 proviral DNA can be detected by Southern blot analysis in most cases of ATLL [76]. Interestingly, extraordinary integration patterns of HTLV-1 proviral DNA are associated with distinct clinical and pathological subtypes and prognosis [77].
Primary cutaneous extranodal NK/T-cell lymphoma, nasal type includes NK-cell lymphomas along with some cytotoxic T-cell lymphomas and lymphomas with indeterminate lineage (NK/T) [78]. These tumors, although rare in Western populations, are not uncommonly seen in Asian and Latin American countries [79–81]. Associated EBV infection is detected in almost all the cases [82, 83]. Patients are present with multiple ulcerated nodules with necrotic centers. The prognosis is poor [84]. Histologically, the lesions show a dense dermal infiltrate of variably sized cells with angiocentric distribution and necrosis [85]. Subcutaneous infiltration by the lymphoma cells, similar to that seen in SPTCL, can be seen. The tumor cells are usually positive for CD56, cytoplasmic CD3, and cytotoxic markers. It is important to demonstrate the presence of EBV, commonly by EBER ISH testing. T-cell receptor (TCR) gene rearrangement is not detected in the true NK-cell neoplasms; however, a subset of cases shows monoclonal TCR gene rearrangements [86, 87]. Assessment of the NK-cell killer immunoglobulin-like receptor (KIR) repertoire through reverse transcriptase PCR has been used to demonstrate monoclonality in these tumors [88].
Another EBV-associated lymphoid proliferation with distinct clinical presentation, regarded in the WHO/EORTC classification as a variant of the previous category, is the hydroa vacciniforme-like cutaneous T-cell lymphoma (HVLL) [89]. It usually affects children and young adults and occurs in regions of Latin America and Asia [90, 91]. Patients present with vesicles and papules on sun-exposed areas that leave scars clinically resemble hydroa vacciniforme. Hypersensitivity to mosquito bites is common. It seems that this disease is a part of the spectrum of EBV-related T-cell lymphoid proliferations of the skin that may include typical hydroa vacciniforme and aggressive lymphomas [92]. The proliferating cells in HVLL usually express CD3, CD2, CD8, and cytotoxic markers. T-cell receptor (TCR) gene rearrangement is detected in a variable proportion of cases [93, 94]. Detection of EBER-1 and BamHI A rightward transcripts (BARTs) by real time PCR from skin crusts and scales of patients with EBV-associated cutaneous lesions has been reported as highly sensitive and specific [95].
Case
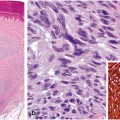
A 38-year-old male presented with multiple papules on chest, neck, and upper extremities. Some of the lesions were crusted with pustule formation. The lesions failed to improve on multiple antibiotic and topical treatments. A biopsy demonstrated dermal lymphocytic infiltrate with minimal epidermotropism. Acute inflammation was present in superficial dermis and epidermis. A dense infiltrate was identified around eccrine coils. This infiltrate was demonstrated to be CD3-positive with some CD4-positive cells involving adnexal structures. Numerous CD8-positive cells were also present. Molecular studies for TCR gamma chain gene rearrangement revealed a clonal peak. A diagnosis of syringotropic mycosis fungoides was made (Fig. 2.6) .
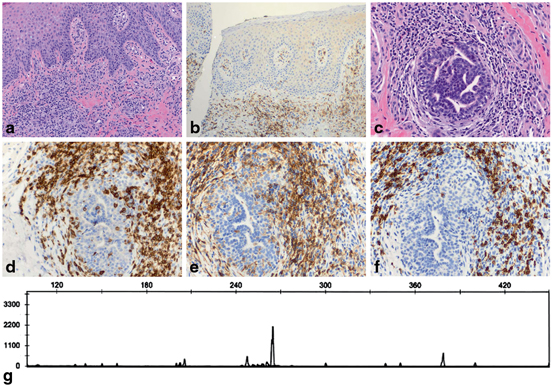
Fig. 2.6
Syringotropic mycosis fungoides. a The lesion shows a dermal infiltrate composed of small lymphocytes with mild cytologic atypia. No epidermotropism is seen. Acute inflammation with intraepidermal neutrophils makes the interpretation more cumbersome (H&E, 10x). b An immunohistochemical study for CD4 highlights only rare small intraepidermal cells (immunoperoxidase, 10x). c Deep dermal lymphocytic infiltrate around eccrine coils. Some hyperchromatic lymphocytes are seen within the adnexal epithelium (H&E, 20x). d A CD3 study highlights the syringotropic T lymphocytes (immunoperoxidase, 20x). e The syringotropic cells express CD4 (immunoperoxidase, 20x). f Numerous CD8-positive T cells are present within the pero-eccrine infiltrate, a confounding finding that may lead to the wrong diagnosis (immunoperoxidase, 20x). g TCR beta chain gene rearrangement analysis reveals a monoclonal peak in a polyclonal background, supporting the diagnosis of syringotropic mycosis fungoides
Comment: Syringotropic mycosis fungoides may be challenging to diagnose in both clinical and histopathological grounds. The lesions seen in this variant of mycosis fungoides are usually multiple papules or nodules, sometimes associated with anhidrosis [96]. Patches or plaques of typical mycosis fungoides are not usually seen. On histology, careful examination is needed to identify the syringotropic cells. In this case, the associated reactive infiltrate composed of neutrophils and reactive CD8-positive T lymphocytes makes the interpretation of the histopathological findings even more difficult. Demonstration of clonality by TCR gene rearrangement studies, along with an adequate interpretation of the pathological and clinical findings, is useful to support a diagnosis of mycosis fungoides in this case. The patient was treated with total skin electron beam therapy with good clinical response.
B-cell Lymphoid Proliferations of the Skin
Primary cutaneous B-cell lymphomas (PCBCL) are B-cell lymphomas originating in the skin with no evidence of extracutaneous disease at the time of diagnosis. The following categories are recognized by the WHO-EORTC (the World Health Organization and the European Organization for Research and Treatment of Cancer)[66, 97]:
1.
Primary cutaneous marginal zone B-cell lymphoma (PCMZL)
2.
Primary cutaneous follicle centre lymphoma (PCFCL)
3.
Primary cutaneous diffuse large B-cell lymphoma, leg type (PCDLBCL, LT)
4.
Primary cutaneous diffuse large B-cell lymphoma, other (PCDLBCL, other)
Besides PCBCL, the skin can be secondarily involved by a variety of nodal or systemic B-cell lymphomas. In fact, the skin is the second most common site of extranodal non-Hodgkin lymphoma following the gastrointestinal tract, with an estimated annual incidence of 1:100,000. Although the different types of PCBCL are histologically similar to their corresponding nodal or systemic counterparts, their clinical behavior and prognosis are often different. For example, nodal diffuse large B-cell lymphoma is considered more aggressive compared to primary cutaneous diffuse large B-cell lymphoma. Therefore, it is crucial to recognize and diagnose PCBCL as separate from nodal or systemic B-cell lymphoma secondarily involving the skin .
Here we discuss the 3 main types of PCBCL: PCMZL, PCFCL, and PCDLBCL, LT, and their differential diagnoses with focus on the utility of molecular testing.
Primary Cutaneous Marginal Zone B-cell Lymphoma
Primary cutaneous marginal zone B-cell lymphoma (PCMZL) is an indolent tumor with almost 100 % 5-year survival [66]. It is composed of small B-cells including marginal zone (centrocyte-like) cells, lymphoplasmacytoid cells, and plasma cells. It is considered a part of the broader mucosa-associated lymphoid tissue (MALT) type lymphomas. It affects adults over 40 years and presents as red to violaceous papules, plaques, or nodules, predominantly on the upper extremities and less often on head and trunk.
Histologically, PCMZL is characterized by nodular to diffuse cellular infiltrate with residual reactive lymphoid follicles. The infiltrating cells comprise predominantly centrocyte-like cells or monocytoid cells of small to medium size with slightly irregular nuclei, moderately dispersed chromatin, inconspicuous nucleoli, and moderately abundant pale cytoplasm. These cells are admixed with variable numbers of lymphoplasmacytoid cells and plasma cells, small numbers of centroblasts and numerous reactive T-cells. Intranuclear (Dutcher bodies) or intracytoplasmic periodic acid-schiff (PAS)-positive inclusions may be present, particularly in cases with predominance of lymphoplasmacytoid cells. Diffuse infiltration involving epithelium and sweat glands and the presence of very immature plasma cells suggests secondary involvement by a systemic lymphoma [98].
Immunophenotypically, the neoplastic lymphocytes express B-cell markers CD19, CD20, CD22, and CD79a, and are negative for CD5, CD10, and CD23. In addition, the neoplastic lymphocytes are BCL2 positive and BCL6 negative. The dendritic cell marker CD21 highlights residual or distorted follicular dendritic cell meshworks, where only rare or absent CD10+ or BCL6+ residual germinal center cells may be found; however, most cells in the infiltrate are CD10− and BCL6−. The lymphoplasmacytoid cells and the plasma cells show monotypic cytoplasmic immunoglobulin light chain expression [99]. If evidence of clonality by light chain restriction is demonstrated by immunohistochemistry, there is no compelling need of performing molecular analysis to demonstrate clonality.
Immunoglobulin heavy chain (IGH) gene rearrangement studies show monoclonal rearrangements in most cases, in the range of 60 % using one set of FR primers, and almost 100 % using three sets of FR primers [23, 100]. Sequence analyses demonstrate somatic hypermutation in the clonally rearranged IGH variable region. Although most cases do not have recurrent chromosomal translocations, a minority of cases show t(14;18)(q32;q21) involving the IGH and MALT1 genes [31] and t(3;14)(p14.1;q32) involving IGH and FOXP1 genes [101]. The t(11;18)(q21;q21) involving the API2 and MALT1 genes that frequently occur in non-cutaneous marginal zone lymphoma of MALT does not occur in PCMZL [102]. Gene rearrangement involving BCL10 has not been reported. Gains of chromosome 3 and 18 may be present [31]. FAS gene mutations are detected in a minority of cases, similar to MZL of other extranodal sites. Mutations of other genes occur rarely and these include the oncogenes PIM1, PAX5, RhoH/TTF, and MYC. Inactivation of the tumor suppressor genes CDKN2A and DAPK by hypermethylation has been documented. These findings suggest different pathogenetic pathways between PCMZL and extracutaneous marginal zone lymphoma of MALT and thus support their distinct classification. It is of interest that cases of PCMZL in Europe, but not in the USA have been associated with Borrelia burgdorferi [97].
Primary Cutaneous Follicle Centre Lymphoma
Primary cutaneous follicle centre lymphoma (PCFCL) is an indolent lymphoma of follicle center B-cells, with a > 95 % 5-year survival [66]. As its indolent behavior, independent of histologic appearance, grading is not recommended for PCFCL. It predominantly affects middle-aged adults, and the sites most frequently affected are trunk, and head and neck regions [103]. It presents as solitary or grouped firm erythematous plaques or nodules. In contrast to PCMZL, multifocal skin lesions are rare.
Histologically, PCFCL shows a spectrum of growth patterns including a follicular, a follicular and diffuse, and a diffuse pattern, with consistent sparing of the epidermis. Follicles are ill-defined with lack of tingible body macrophages and attenuated or absent mantle zones. The abnormal follicles are composed of a mixture of centrocytes, relatively few centroblasts, and many reactive T-cells, enmeshed in a network of follicular dendritic cells. In advanced lesions, the follicular structures are no longer discernible, and the infiltrating cells are generally large centrocytes with variable admixture of centroblasts and immunoblasts .
Immunophenotypically, the neoplastic lymphocytes express B-cell markers, with co-expression of the germinal center marker BCL6. CD10 is detected in cases with follicular growth pattern but is uncommon in cases with diffuse growth pattern. Focal positivity for MUM-1 is possible, although the tumor cells are usually negative [104]. The neoplastic cells are negative for CD5 and CD43. Unlike nodal and secondary follicular lymphomas, PCFCL cells generally do not express or are faintly positive for BCL2. A strong expression of BCL2 suggests secondary skin infiltration. Monotypic immunoglobulins can be detected by flow cytometry or frozen sections but rarely by routine paraffin immunohistochemistry.
Molecular testing reveals monoclonal IgH gene rearrangements in 67 % of the cases. Somatic hypermutations of the variable IgH gene are common, thus the chance of false negatives can be decreased with the use of multiple primers sets [23, 100]. In contrast to systemic follicular lymphoma, PCFCL commonly lack the t(14;18)(q32;q21) or BCL2 gene rearrangement [105, 106]. Mutations of oncogenes BCL6, MYC, RhoH/TTF, and PAX5 have been reported. Inactivation of the CDKN2A tumor suppressor gene by deletion or promoter hypermethylation is rare [107]. Array-based comparative genomic hybridization (aCGH) and FISH studies may rarely reveal amplification of chromosome region 2p16 (containing the c-REL and BCL11A genes) [108] and loss of chromosome 14q32 (containing the IGH gene) [107].
Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type
Primary cutaneous diffuse large B-cell lymphoma, leg type (PCDLBCL, LT) is a subtype of diffuse large B-cell lymphoma with an aggressive behavior, and histologically is composed of large transformed cells with a diffuse growth pattern. Primary cutaneous diffuse large B-cell lymphoma, leg type (PCDLBCL, LT) characteristically involves the legs, [109] but sometimes can occur in trunk and rarely in the head. Primary cutaneous diffuse large B-cell lymphoma, leg type (PCDLBCL, LT) predominantly affects elderly females and presents as multiple rapidly growing nodules [110]. In contrast to indolent cutaneous B-cell lymphomas, they more often disseminate to extracutaneous sites and have a poorer prognosis.
Histologically, PCDLBCL, LT, shows a diffuse infiltrate of large cells in the dermis with a destructive growth pattern obliterating adnexal structures and extending into the subcutaneous tissue, with sparing of the epidermis. The infiltrating tumor cells monotonous population or confluent sheets of centroblasts and immunoblasts [111]. Mitotic figures are frequently observed.
Immunophenotypically, the neoplastic lymphocytes express B-cell markers, and light chain restriction can be demonstrated by flow cytometry immunophenotype or frozen section, but no paraffin section immunohistochemistry. The neoplastic cells commonly express BCL2 and IRF4/MUM1 [112, 113]. They variably express BCL6 and generally do not express CD10 .
Molecular testing reveals monoclonal IgH gene rearrangements in almost 100 % of cases [23]. The t(14;18) involving the BCL2 gene is usually not present [114], however, cases with gene amplification of BCL2 have been reported [115]. Gene expression profiling studies showed gene expression profile similar to that of activated B-cell type of nodal or systemic DLBCL [116]. Array-based comparative genomic hybridization (aCGH) and FISH studies showed frequent deletion of chromosome 9p21 (containing the CDKN2A and CDKN2B genes) and amplification of 18q21 (containing the BCL2 and MALT1 genes). Other chromosomal aberrations include gains in chromosomes 1, 2, 3, 7, 12, and 18q, and losses in 6q, 13, and 17 [108]. Inactivation of CDKN2A by promoter hypermethylation is also detected. The inactivation of CDKN2A, either by deletion or promoter hypermethylation is a poor prognostic marker [107].
Case
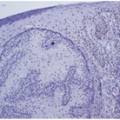
A 54-year-old male presented with nodules in the scalp. The patient did not have a significant medical history. On physical examination, scalp nodules, less than 1 cm in diameter were palpated, and a punch biopsy was obtained. The histologic sections showed a dense dermal infiltrate without epidermal involvement. There were scattered lymphoid follicles with germinal center formation. Immunohistochemical studies showed the lymphoid follicles to react with B-cell markers CD20 and PAX-5. BCL2 was negative in the germinal centers (Fig. 2.7 a and b).
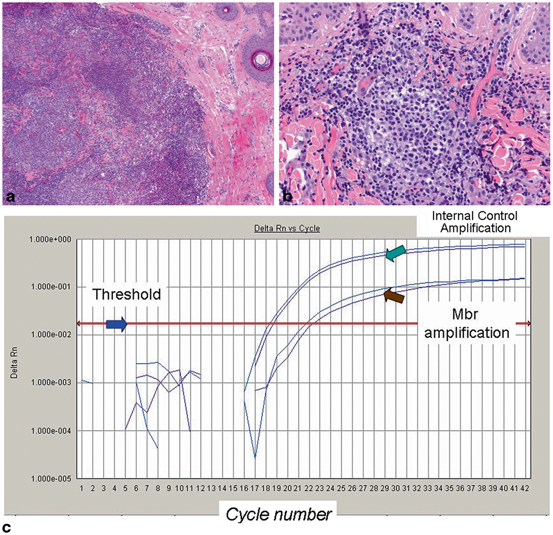
Fig. 2.7
Primary cutaneous follicle centre lymphoma harboring the t(14:18)(q32;q21) IGH/BCL2 translocation. a The skin biopsy shows a dense dermal infiltrate with ill defined follicles with germinal centers (H&E, 10x). b The germinal centers are composed of a mixture of centrocytes, centroblasts, and many reactive small lymphocytes (H&E, 20x) c Molecular analysis for IGH-BCL2 rearrangement associated with the t(14;18)(q32;q21.3) was performed by real-time PCR using specific primers that target the major breakpoint region (mbr) of the BCL2 gene. Amplification of the cyclophilin gene is used as an internal control for sample adequacy for PCR. IGH gene rearrangement analysis revealed a polyclonal pattern (not shown). Although the presence of IGH-BCL2 translocation should raise suspicion for systemic follicular lymphoma with secondary involvement of the skin, the clinical workup in this patient was negative for systemic disease and a diagnosis of primary cutaneous follicle centre lymphoma was favored
Comment: The differential diagnosis is between follicular lymphoid hyperplasia and primary cutaneous follicular lymphoma. Molecular testing for IGH gene rearrangements yielded a polyclonal pattern, thus it failed to detected monoclonality. However, because of the possibility of follicular lymphoma, testing for t(14:18)(q32;q21) IGH/BCL2 by PCR was performed and demonstrated the presence of the rearrangement (Fig. 2.7c). This rearrangement is commonly associated with nodal follicular lymphoma, but it can also be present in some primary cutaneous follicular lymphomas, as illustrated in this case. On follow up, clinical staging was negative, and the disease was confined to the scalp. Local radiation, but no systemic therapy was administered, and the patient was asymptomatic on last follow up 5 years after diagnosis .
Leukemia Cutis
Leukemia cutis (LC) is an all encompassing term which describes a neoplastic infiltrate of the skin by myeloid (granulocytic or monocytic) or lymphoid precursor cells. LC composed of myeloid blast with granulocytic differentiation may also be designated as chloroma, granulocytic sarcoma, extramedullary myeloid sarcoma and monoblastic sarcoma if precursor cells demonstrate monoblast or promonocytic differentiation [117, 118]. LC may be present in the context of known myeloid or lymphoid disorders or may present as aleukemic LC in patients with no prior history of a hematologic malignancy. Aleukemic LC occurs in majority of cases which demonstrate myelomonocytic or monocytic differentiation [119–121].
Molecular analysis of LC lesions has been reported for a variety of hematopoietic malignancy including AML, acute promyelocytic leukemia, and mature T-cell leukemias [122–126]. Although, genetic abnormalities in LC lesions appear to be concordant with testing in bone marrow there are some exceptions and molecular testing may be beneficial in aleukemic LC, particularly if the infiltrate is composed of myeloid blasts since recurring genetic abnormalities defines some categories of AML classification. The 2008 World Health Organization (WHO) classification scheme of AML incorporates clinical, immunophenotypic and cytogenetic features which classifies AML in four main categories: (1) AML with recurrent genetic abnormalities, (2) AML with myelodysplasia related changes, (3) AML related to therapy, and (4) AML not otherwise specified [117




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
