Joseph M. Lam, Li-Chuen Wong This chapter covers neonatal hair patterns, genetic hair shaft abnormalities, and the conditions in which hypo- or hypertrichosis are present in the neonatal period. There are many syndromes in which hypotrichosis or atrichia occur, and those in which it is a prominent feature are discussed. Localized alopecia can occur physiologically, with trauma, and as a nevoid disorder, either alone or associated with other nevi. Diffuse hypertrichosis can occur alone or as part of various syndromes. Localized hypertrichosis may occur with other nevi, but also may be a marker for serious neural tube closure defects (see also Chapter 9). The hair root is characterized by three definable stages of growth: anagen, catagen, and telogen. The anagen stage is a growth phase that may last for many years. The catagen stage is a brief transition from the growth phase (anagen) to an inactive state. The telogen stage is the resting phase following catagen and lasts approximately 3 months. In adults, each region of the scalp contains hairs in different phases of the hair cycle. In the fetus, hair follicles appear at about 10 weeks’ gestation and by 20 weeks’ gestation, all the hair on the scalp is in anagen phase. At about 30 weeks’ gestation, the hair in the frontal and parietal region rapidly transition through a catagen and telogen phase, representing the first shedding for the fetus. At or just before birth, scalp hair grows in synchronized waves, starting with the hairs in the frontal and parietal regions and ending with the occipital region. Hair density, structure, and growth vary depending on sex, ethnicity and nutritional status. From the 18th week after birth, changes in growth patterns progressively adopt the dyssynchronous pattern of the adult scalp.1–3 The sloping angle of scalp hair results from stretch on the scalp from the growth of the underlying fetal brain during weeks 10–16 of gestation. The usual location of the parietal hair whorl is several centimeters anterior to the posterior fontanelle. Of normal Caucasian infants, 95–98% have a single hair whorl in the parietal area,4,5 usually clockwise but inconsistent in position. The remainder have a double parietal whorl. Only 10% of African-American individuals with short curly hair have a parietal whorl.5 A mild frontal upsweep or ‘cowlick’ is present in 7% of normal infants.4 Hair patterns may be very abnormal in infants with structural abnormalities of the brain, demonstrating a striking frontal upsweep and absent or aberrant parietal whorls.4 Multiple parietal whorls occur with increased frequency in developmentally delayed children, and their presence in the neonate may be an early sign of such a disorder.6 A recent study of hair whorl orientation and handedness suggests that a single gene controls these traits.7 The frontal hairline of neonates is lower than in older children, a feature most striking in racial groups in which there is profuse hair at birth. These terminal hairs on the brow are gradually replaced over the first 12 months of life by vellus hairs. Normally, posterior hairline hair roots are located above the neck crease. Low frontal and posterior hairlines are each associated with several syndromes, summarized in Box 31.1. Heterochromia of the hair is described as the growth of hair with two distinct colors in the same person.8 In piebaldism, there is a white forelock, which will be obvious in a dark-haired neonate. A congenital melanocytic nevus may present as a tuft of dark hair, which is often also longer and coarser than the surrounding normal hair. In hereditary, usually autosomal dominant, heterochromia, there may be a tuft of red hair in a dark-haired neonate or a dark tuft in a fair individual. There has recently been a report of a diffuse heterochromia of scalp hair, present from birth, with black and red hairs evenly distributed over the scalp8 and heterochromia of the scalp hair following Blaschko’s lines.9 A diverse group of conditions can result in hair shaft abnormalities. Some are associated with extracutaneous disease, whereas others affect only the hair itself. These conditions have been reviewed in detail by Whiting,10 Price,11 and Rogers.12,13 Trichoscopy performed with a handheld dermoscope or a video-dermoscope is becoming a useful tool in the diagnosis of hair shaft disorders.14 Monilethrix is a condition that produces a beaded appearance of the hair and the term comes from the Latin word for necklace (monile) and the Greek for hair (thrix).10–12 Most families with monilethrix show an autosomal dominant inheritance pattern, but autosomal recessive forms also exist. The hair is usually normal at birth but is replaced within weeks by affected hairs that are dry, dull, and brittle, breaking spontaneously and leaving a stubble-like appearance (Fig. 31.1). The hairs may break almost flush with the scalp or may attain lengths of 0.5–2.5 cm, or occasionally longer. There may be spontaneous improvement with time, especially during puberty and pregnancy, but the condition never resolves completely. Follicular keratosis is commonly associated and may involve the scalp, face, and limbs. On microscopy, spindle-shaped ‘nodes’ separated by constricted internodes are seen (Fig. 31.2 This is characterized by groups of three or four regularly spaced twists of the hair shaft on its own axis (Fig. 31.3 Pili torti can occur alone or as a manifestation of defined syndromes, some of which are identifiable in the neonatal period. Menkes syndrome is an X-linked recessive condition caused by mutations in a gene encoding for a protein believed to be a copper-transporting P-type ATP-ase,17 and the multiple abnormalities are due to decreased bioavailability of copper, with resultant functional deficiencies of copper-dependent enzymes. In the early months of life, scalp and eyebrow hair becomes kinky, coarse, and sparse. Lax, pale skin, hypotonia, and early neurodegenerative changes may already be seen in the neonatal period. In Bazex syndrome, inherited as an X-linked dominant trait,18 congenital hypotrichosis with pili torti is associated with follicular atrophoderma and multiple facial milia, both of which may also be present from birth. These patients have an increased susceptibility to the development of basal cell carcinomas. In Björnstad syndrome, pili torti is associated with sensorineural deafness and occasionally mental retardation.19 In later childhood, normal hair may replace the affected hairs, with a considerable improvement in appearance. Both autosomal dominant and recessive inheritance patterns have been reported. Recently the gene for Björnstad syndrome has been mapped to chromosome 2 in a family with an autosomal recessive mode of inheritance (Fig. 31.5 The term trichorrhexis nodosa (TN) refers to the light microscopic appearance of a fracture with splaying out and release of individual cortical cells from the main body of the hair shaft, producing an appearance suggestive of the ends of two brushes pushed together (Fig. 31.6 There is a growing list of conditions associated with trichorrhexis nodosa (Box 31.2). The term ‘trichothiodystrophy’ refers to the sulfur-deficient brittle hair that is a marker for a neuroectodermal symptom complex occurring in a group of autosomal recessive genetic disorders.22 There is considerable genetic heterogeneity in trichothiodystrophy.23 Named syndromes that fit into this spectrum include: Tay, Pollitt, Sabinas brittle hair, and Marinesco–Sjögren syndromes. The major clinical features seen in this group of conditions are photosensitivity with a DNA repair defect (due to mutations in the XPD ECCR2 DNA repair/transcription gene), ichthyosis, brittle hair, intellectual impairment, decreased fertility, short stature,24–26 and osteosclerosis. Some authors use mnemonic acronyms including PIBIDS, IBIDS, and BIDS to identify patients by these key clinical characteristics (see also Chapter 19).23 Features that may be evident in the neonatal period are intrauterine growth retardation, severe infections, congenital cataracts, nail dystrophy, facial dysmorphism, a collodion baby phenotype, and the characteristic fragile, dull, short, disordered hair involving the scalp hair, eyebrows, and eyelashes (Fig. 31.7).27,28 On light microscopy the hair has a wavy, irregular outline and a flattened shaft, in which twists like a folded ribbon occur. Two types of fracture are seen: an atypical trichorrhexis nodosa and trichoschisis, a clean, transverse fracture (Fig. 31.8A Woolly hair (WH) is an abnormal variant of tightly curled hair. Compared with normal curly hair, WH does not grow well and stops growing at a few inches. Several families have been reported in which some affected individuals exhibit features of hypotrichosis while others have woolly scalp hair.30 Clinically, WH can be divided into syndromic and non-syndromic forms. The syndromic form includes Naxos disease, cardiofaciocutaneous syndrome and Carvajal syndrome. Naxos disease is an autosomal recessive disorder that combines palmoplantar keratoderma and other ectodermal features with arrhythmogenic right ventricular dysplasia/cardiomyopathy. It was first reported in families on the Greek island of Naxos and is associated with a deletion in the plakoglobin gene. Cardiofaciocutaneous (CFC) syndrome is characterized by a distinctive facial appearance, heart defects, and mental retardation and is caused by heterozygous gain-of-function mutations in one of four different genes: KRAS, BRAF, MEK1, or MEK2. Some patients have associated woolly hair. In Carvajal syndrome, patients present with epidermolytic palmoplantar keratoderma, woolly hair, and dilated cardiomyopathy. Carvajal syndrome can be caused by mutations in the gene encoding desmoplakin.31–36 Non-syndromic forms of hereditary woolly hair present as autosomal dominant (ADWH) or autosomal recessive (ARWH). Dominant forms of WH have been linked to mutations in the helix initiation motif of KRT74. Recessive forms of WH have been linked to mutations in the LIPH and the LPAR6/P2RY5 genes, both expressed in the inner root sheath of the hair follicle. Localized and sporadic woolly hair (the woolly hair nevus) (Fig. 31.9) can also occur. Uncombable hair is a relatively rare anomaly of the hair shaft that results in a disorganized, unruly hair pattern that is impossible to comb flat.11,13,37 Synonyms are spun-glass hair, pili canaliculi, and pili trianguli et canaliculi. In the classic clinical form, the hair is a light silvery-blond, paler than expected. It is frizzy, stands away from the scalp, and cannot be combed flat. It is often ‘spangled’ or glistening (Fig. 31.10). It is usually normal in length, quantity, and tensile strength. The onset may be with the first terminal growth or later. Eyebrows, lashes, and body hair are normal. Most cases improve with the onset of puberty. There are reports suggesting both dominant and recessive inheritance patterns. Scanning electron microscopy best demonstrates the characteristic shallow grooving or flattening of the surface.37 These areas are often discontinuous and change orientation many times along the length of the hair, occurring on different planes of the hair at different points. Cross-sectional microscopy shows triangular, reniform, and other unusual shapes. Most children with uncombable hair are otherwise normal; the findings have been demonstrated in a variety of other syndromes with congenital onset, including progeria, Marie Unna hypotrichosis,38 Rapp–Hodgkin syndrome,39 orofacial digital syndrome type I,40 ectrodactyly ectodermal dysplasia and clefting syndrome,40 and hypohidrotic ectodermal dysplasia.39 The classic clinical appearance of spun-glass or uncombable hair would seem to depend on the proportion of abnormal hairs. This hair shaft abnormality, which may be present at birth, does not result in significant hair fragility. The hair looks pleasantly shiny, and on close observation alternating bright and dark bands are seen.41 There are usually no associated abnormalities. The condition may be sporadic or inherited, usually as a dominant characteristic. The bright areas are due to light scattered from clusters of air-filled cavities within the cortex, and in a hair mount, viewed with transmitted light, the light areas appear as dark patches. Scanning electron microscopy shows longitudinal wrinkling and folding in bands corresponding to the abnormal areas, possibly due to the evaporation of air in the spaces when the hair is coated in the vacuum. Transmission electron microscopy demonstrates multiple holes within the cortex. Recently, a locus for pili annulati was mapped to Ch12q24.32–24.33.42 This is the characteristic hair shaft abnormality of Netherton syndrome, an autosomal recessive condition due to mutations in the SPINK5 gene, which encodes for the serine protease inhibitor LEKTI (see also Chapter 19).43 Although the severity varies considerably, the clinical and microscopic findings are present from birth. In the severely affected neonate, the hair may be extremely sparse or even absent altogether (Fig. 31.11). What hair is present is short and dull and breaks easily. The changes may affect eyebrows, eyelashes, and general body hair.13 Hair mount demonstrates a ball-and-socket configuration with various patterns seen (Fig. 31.12 As discussed in the previous section, many hair shaft abnormalities present in the neonatal period or in early infancy with significant hypotrichosis. There appear to be several distinct genotypes within this group, with recessive, dominant, and X-linked inheritance patterns being represented.46,47 Those with recessive inheritance are in general the most severe and congenital in onset. In some pedigrees, there is a total absence of hair (congenital atrichia, atrichia congenita, alopecia universalis congenita) and on biopsy no hair follicles are found. Mutations in the hairless gene on chromosome 8 have been demonstrated in some families.46 In some dominant pedigrees, the hair is present but extremely sparse (congenital hypotrichosis, hypotrichosis simplex), with biopsy demonstrating a few scattered, miniaturized follicles occurring in decreased numbers; only the scalp is involved, the hair elsewhere being normal. In some of these pedigrees mutations have been found in corneodesmosin, a keratinocyte adhesion molecule.48 The hair in this autosomal dominant condition is usually sparse or absent at birth, but it is not until early childhood that the characteristic coarse, wiry hair appears, showing flattening and irregularly distributed twisting on microscopy.49,50 The condition has been mapped to a mutation in the inhibitory upstream open reading frame of the hairless gene on chromosome 8p21.51,52 This is a distinctive association of congenital atrichia and tiny, white papules.53 Atrichia of the scalp may be present from birth or appear in early childhood. In most cases, fetal hair is shed in the first 3 months of life and never replaced; eyebrows and eyelashes may or may not be involved (Fig. 31.13). The papular lesions, which occur diffusely but predominate on the face and scalp, are not present in the neonatal period. Histopathology shows the papules to represent keratin-filled follicular cysts in contact with the overlying epidermis. Recent work has demonstrated mutations in the hairless gene on chromosome 8, as seen also in alopecia universalis congenita.47 Vitamin D-dependent rickets type 2A (VDDR2A) can present with alopecia that is clinically and pathologically indistinguishable from that seen in atrichia with papular lesions.54 In this condition, which bears some clinical similarity to atrichia with papular lesions, there is hypotrichosis with sparse, coarse hair, and multiple milia are present at birth on the face and sometimes also the limbs and trunk. Study of a large pedigree suggests X-linked dominant inheritance.55 Hair that is short and sparse from birth is a feature of this rare autosomal recessive disorder in which progressive macular degeneration can lead to blindness during the first to fourth decades. The hair may be morphologically normal or show a variety of nonspecific shaft abnormalities. The disease results from mutations in CDH3 encoding P-cadherin.56 Congenital hypotrichosis is a feature of this condition, accompanied later in life by lymphedema and telangiectasia. Mutations have been found in the transcription factor gene SOX18.57 Hypotrichosis (see also Chapter 29) is an important feature in many ectodermal dysplasias,58 but often becomes obvious only after the neonatal period. A selection of conditions in which there may be congenital or early-onset severe hypotrichosis or atrichia will be considered here. Hypotrichosis of a variable and sometimes very severe degree of scalp hair, eyebrows, eyelashes, and body hair is usually present at birth.59 Any hair present is fine and fragile. Later significant features are leukoplakia, nail dystrophy, and palmoplantar keratoderma. The condition is caused by mutations in GJB6, coding connexin 30.59 Rare cases of Clouston syndrome have sensorineural deafness which may represent a contiguous gene syndrome resulting from deletion of the GJB6 gene and of the connexin-26 gene (GJB2).60 This condition may be inherited due to one of four defects: (1) the EDA gene, which encodes ectodysplasin-A on Xq13.1, leading to X-linked hypohidrotic ectodermal dysplasia (HED); (2) the ectodysplasin receptor (EDAR) on 2q12.3, with autosomal dominant and recessive forms; (3) the EDAR death domain (EDARADD) on 1q42-q43, with autosomal dominant and recessive forms; (4) the NEMO gene on Xq28, which presents with immunodeficiency, and occasionally osteopetrosis and lymphedema.61 The majority of cases represent a mutation in the EDA gene and are characterized by marked hypotrichosis of all hair-bearing areas, often evident in the neonatal period; hair that is present is fine and fair, and often shows pili canaliculi on microscopy. Other features that may be evident in the neonatal period include impaired heat regulation, diffuse scaling of skin, hypoplastic or absent nipples, and the typical facies with a depressed nasal bridge and prominent brow.62 Rouse and colleages63 reported that scalp biopsies from patients with hypohidrotic ectodermal dysplasia (HED) demonstrate an absence of eccrine structures in the majority of cases. Their absence is diagnostic of HED, and their presence suggests that the patient does not have this disorder. As the eccrine apparatus is fully formed by the third trimester this test should be reliable in the neonate.63 At birth in the ankyloblepharon, ectodermal dysplasia and clefting syndrome (AEC, Hay–Wells), the scalp is usually red and scaly with extensive erosions and crusts, and there is a severe hypotrichosis (Fig. 31.14).64 Other neonatal features include a generalized erythroderma with or without erosive lesions, ankyloblepharon filiforme, lacrimal duct atresia, cleft palate and lip, and hypoplastic nails. Both Rapp–Hodgkin and AEC syndromes have mutations in 3q27 which encodes the p63 gene. As such, they are currently viewed as related disorders, with reported cases of Rapp–Hodgkin syndrome sharing all the features of AEC apart from the ankyloblepharon.58,65 The main features of this probably X-linked dominant condition are congenital hypotrichosis, milia with onset in the first 3 months of life, the later appearance of follicular atrophoderma as the milia are shed, and early development of basal cell carcinomas.18 Microscopic hair shaft examination may show trichorrhexis nodosa and an irregular twisting. In this condition, after the shedding in the first weeks of life of an initial sparse cover of hair, there is almost total alopecia with only tiny vellus hairs being evident; scalp biopsy demonstrates atrophy of hair follicles and rudimentary hair shafts.66 Nail dystrophy and an abnormal facies with a broad nasal bridge, hypertelorism, a broad nose, and a long philtrum are other congenital features. In this group of conditions (see also Chapter 19), which includes autosomal recessive and autosomal dominant forms of lamellar ichthyosis, congenital ichthyosiform erythroderma, and lamellar ichthyosis of the newborn (self-healing collodion baby), the hair is often either absent or shed in the early weeks of life with the collodion scale (Fig. 31.15). This combination of traits has been described as a new autosomal recessive genodermatosis.67 Hypotrichosis of scalp, eyebrows, and eyelashes is evident in the neonatal period, and the ichthyosis and follicular atrophoderma are both also congenital. Woolly hair was an additional feature in one case.67 Severe hypotrichosis of scalp, eyebrows, and eyelashes may be evident at birth (Fig. 31.16). Other congenital features include spiny follicular plugs, perioral furrowing, reticulate hyperkeratosis of the palms and soles, widespread thickened erythematous plaques, and hearing loss. The condition is usually caused by mutations in GJB2, encoding connexin 26.48,59 From birth these individuals demonstrate keratotic follicular papules, atrichia, or severe hypotrichosis and photophobia.68 A variety of other features can be present and there are pedigrees suggesting both X-linked recessive and autosomal dominant inheritance. Happle69 suggests there may be more than one syndrome within this designation. A functional deficiency of a zinc metalloprotease causes the disease (MBTPS2).70 Hereditary mucoepithelial dysplasia is a dominantly inherited disease characterized by congenital nonscarring hypotrichosis with coarse abnormal hair, gingival erythema, severe keratitis, follicular keratotic papules, and periorificial psoriasiform plaques.71 It has been suggested that this and IFAP may be the same condition, but there are sufficiently different features to make it likely they are separate entities. Searches for abnormal expression of gap junction and desmosomal proteins have so far been unrewarding.71
Hair Disorders
Introduction
Neonatal hair development
Scalp hair whorls
The hairline
Heterochromia of scalp hair
Hair shaft abnormalities
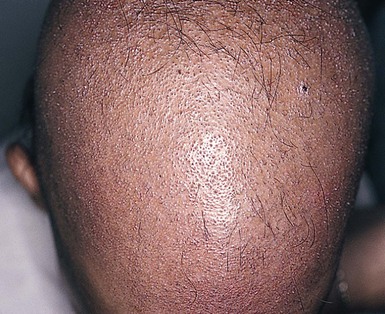
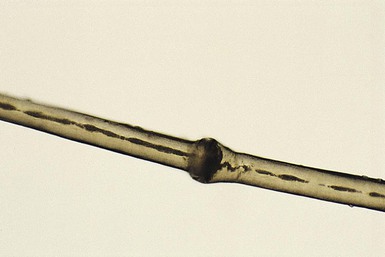
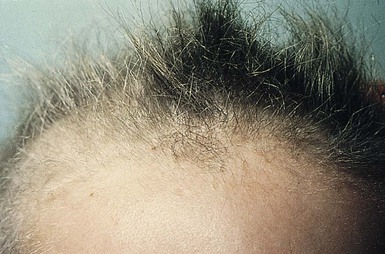
Monilethrix
 ). The nodes have the diameter of normal hair and may be medullated, whereas the internodes are narrower and usually nonmedullated, and are the sites of fracture. In monilethrix the hair shaft fragility is due to structural weakness of the hair fiber that is caused by a genetic defect in keratin intermediate filament protein. Mutations in the hair cortex keratin genes KRT81 (hHB1), KRT83 (hHB3), and KRT86 (hHB6) have been identified in autosomal dominant monilethrix.15 An autosomal recessive form of monilethrix is caused by mutation in the DSG4 gene and presents with more extensive alopecia of the scalp, body, and limbs, and a papular rash involving the extremities and periumbilical region.16
). The nodes have the diameter of normal hair and may be medullated, whereas the internodes are narrower and usually nonmedullated, and are the sites of fracture. In monilethrix the hair shaft fragility is due to structural weakness of the hair fiber that is caused by a genetic defect in keratin intermediate filament protein. Mutations in the hair cortex keratin genes KRT81 (hHB1), KRT83 (hHB3), and KRT86 (hHB6) have been identified in autosomal dominant monilethrix.15 An autosomal recessive form of monilethrix is caused by mutation in the DSG4 gene and presents with more extensive alopecia of the scalp, body, and limbs, and a papular rash involving the extremities and periumbilical region.16

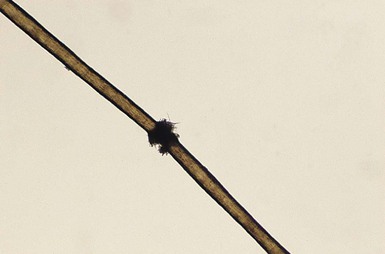
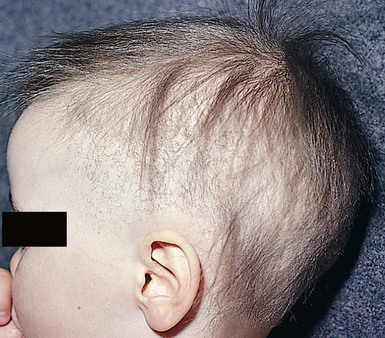
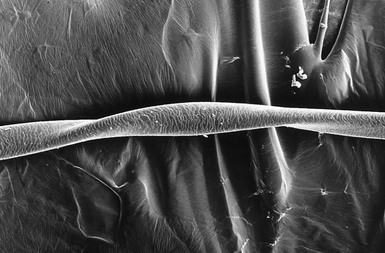
Pili torti
 ).9,10 Microscopically, twists are seen, each 0.4–0.9 mm in width, occurring usually in groups of three or more at irregular intervals. Twists are almost always 180°, although some are 90° or 360°. The hair shaft is somewhat flattened. Pili torti may occur as an isolated phenomenon, with onset at birth or in the early months of life. The hair is usually fairer than expected and is spangled, dry, and brittle, breaking at different lengths (Fig. 31.4). It may stand out from the scalp and tends to be short, especially in areas subject to trauma.
).9,10 Microscopically, twists are seen, each 0.4–0.9 mm in width, occurring usually in groups of three or more at irregular intervals. Twists are almost always 180°, although some are 90° or 360°. The hair shaft is somewhat flattened. Pili torti may occur as an isolated phenomenon, with onset at birth or in the early months of life. The hair is usually fairer than expected and is spangled, dry, and brittle, breaking at different lengths (Fig. 31.4). It may stand out from the scalp and tends to be short, especially in areas subject to trauma.
 ).19 Pili torti may occur also in Rapp–Hodgkin syndrome, although pili canaliculi is the more characteristic finding.
).19 Pili torti may occur also in Rapp–Hodgkin syndrome, although pili canaliculi is the more characteristic finding.
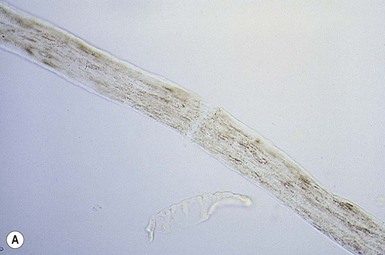
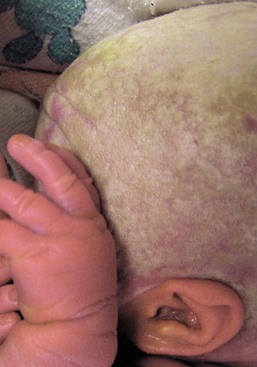
Trichorrhexis nodosa
 ).10–12 When the break occurs, the brush-like end is clearly seen. Electron microscopy shows the disrupted cuticle and splaying of cortical cells. The defect renders the hair very fragile, and it breaks readily with trauma – or sometimes probably spontaneously. In congenital autosomal dominant TN, the hair is usually normal at birth but is replaced within a few months with abnormal, fragile hair. Trichorrhexis nodosa was found in eight of 25 children with mitochondrial disorders in the absence of skin manifestations, suggesting that hair examination may be a useful diagnostic tool when these disorders are suspected.20 Trichorrhexis nodosa is also seen in trichohepatoenteric syndrome, a condition that includes intractable diarrhea of infancy, facial dysmorphism, developmental delay, and immunodepression.21
).10–12 When the break occurs, the brush-like end is clearly seen. Electron microscopy shows the disrupted cuticle and splaying of cortical cells. The defect renders the hair very fragile, and it breaks readily with trauma – or sometimes probably spontaneously. In congenital autosomal dominant TN, the hair is usually normal at birth but is replaced within a few months with abnormal, fragile hair. Trichorrhexis nodosa was found in eight of 25 children with mitochondrial disorders in the absence of skin manifestations, suggesting that hair examination may be a useful diagnostic tool when these disorders are suspected.20 Trichorrhexis nodosa is also seen in trichohepatoenteric syndrome, a condition that includes intractable diarrhea of infancy, facial dysmorphism, developmental delay, and immunodepression.21
Trichothiodystrophy
 ). Using crossed polarizers, light and dark bands are seen when the hair is aligned in one of the polarizer directions – the so-called tiger-tail appearance (Fig. 31.8B
). Using crossed polarizers, light and dark bands are seen when the hair is aligned in one of the polarizer directions – the so-called tiger-tail appearance (Fig. 31.8B ). This may be absent at birth and is not fully developed until 3 months of age.29 Scanning electron microscopy shows irregular ridging and fluting and a disordered, reduced, or absent cuticle scale pattern.
). This may be absent at birth and is not fully developed until 3 months of age.29 Scanning electron microscopy shows irregular ridging and fluting and a disordered, reduced, or absent cuticle scale pattern.
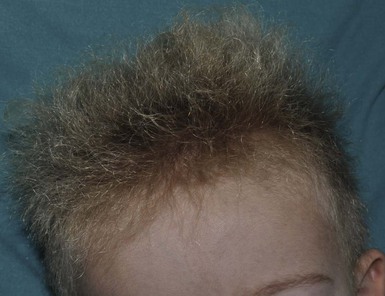
Woolly hair
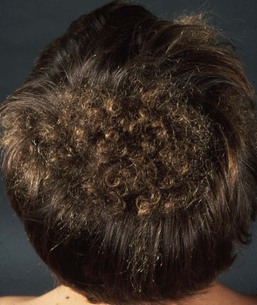
Uncombable hair
Pili annulati
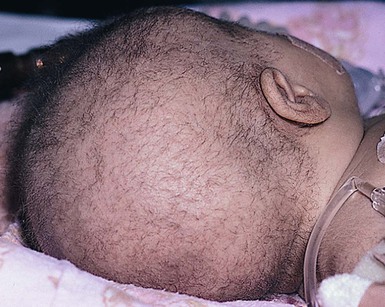
Trichorrhexis invaginata
 ). The classic ‘bamboo hair’ occurs when the soft abnormal hair shaft wraps around a firmer distal shaft, producing the appearance of a shallow invagination of the distal into the proximal shaft. There is a tulip-like form with a deeper invagination and longer sides of the ‘cup.’11 Circumferential strictures may be found, representing the earliest stage of the invagination. The term ‘golf-tee hair’ has been given to the expanded proximal end of an invaginate node after a break has occurred. Thin vellus hairs may show multiple invaginations, the so-called ‘canestick hairs.’44 A helical pattern of twisting with obliquely running parallel invaginations has recently been described.45
). The classic ‘bamboo hair’ occurs when the soft abnormal hair shaft wraps around a firmer distal shaft, producing the appearance of a shallow invagination of the distal into the proximal shaft. There is a tulip-like form with a deeper invagination and longer sides of the ‘cup.’11 Circumferential strictures may be found, representing the earliest stage of the invagination. The term ‘golf-tee hair’ has been given to the expanded proximal end of an invaginate node after a break has occurred. Thin vellus hairs may show multiple invaginations, the so-called ‘canestick hairs.’44 A helical pattern of twisting with obliquely running parallel invaginations has recently been described.45
Diffuse alopecia (hypotrichosis)(Box 31.3)
Hypotrichosis with hair shaft abnormalities
Isolated congenital alopecia or hypotrichosis without other defects
Marie-unna hypotrichosis
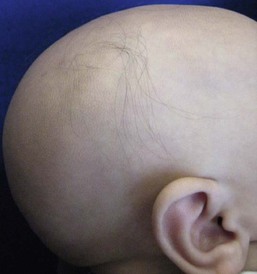
Atrichia with papular lesions
Congenital hypotrichosis and milia
Hypotrichosis with juvenile macular dystrophy
Hypotrichosis–lymphedema–telangiectasia syndrome
Hypotrichosis associated with ectodermal dysplasias
Hidrotic ectodermal dysplasia (Clouston syndrome)
Hypohidrotic ectodermal dysplasia
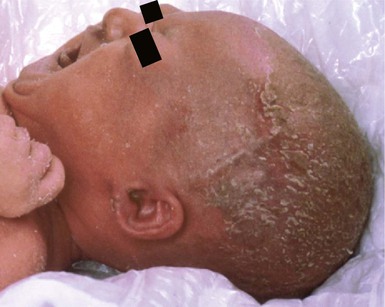
Ankyloblepharon, ectodermal dysplasia and clefting syndrome, and Rapp–Hodgkin syndrome
Bazex–Dupré–Christol syndrome
Congenital atrichia with nail dystrophy, abnormal facies, and retarded psychomotor development
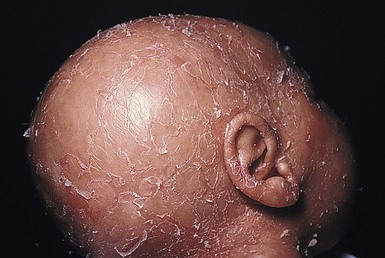
Hypotrichosis associated with ichthyoses
Ichthyoses presenting as the collodion baby phenotype
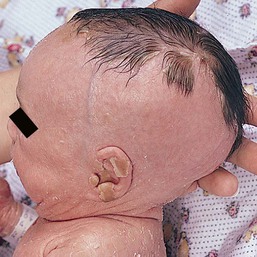
Congenital ichthyosis, follicular atrophoderma, hypotrichosis, and hypohidrosis
Keratitis, ichthyosis, and deafness (KID) syndrome
Ichthyosis follicularis, congenital atrichia, and photophobia (IFAP)
Hypotrichosis with hereditary mucoepithelial dysplasia
Hypotrichosis with premature aging syndromes
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Hair Disorders
31