Peptide
Sites of synthesis
Stimulus
Actions
Mediation of action
Molecular forms
Anorexigenic
OXM
L cells of distal ileum and colon
Pancreas
CNS
Meal
Calorie content
Fat
Inhibits food intake
Inhibits gastric acid secretion
Inhibits gastric motility
Reduces pancreatic enzyme secretion
GLP-1 receptor
Glucagon receptor
Suppression of ghrelin
–
GLP-1
L cells of distal small ileum, colon
Pancreas
CNS
Meal
Incretin effect on insulin secretion
Suppresses glucagon release
Promotes pancreatic β-cell growth
Inhibits food intake
Delays gastric emptying
Inhibits gastric secretion
Inhibits lipase secretion
GLP-1 receptor
GLP-17-36
GLP 17-37
PYY3-36
L cells of distal ileum, colon, rectum
CNS
Meal
Fat and protein
Calorie content
CCK, Gastric acid, Bile acid, Bombesin,
IGF-1
Inhibits food intake
Reduces gastric motility
Inhibits gallbladder secretion
Inhibits pancreatic secretion
Y2 receptor
Inhibits NPY
PYY 1-36
PYY 3-36
PP
PP cells in islets of Langerhans
CNS
Inhibits pancreatic enzyme secretion
Inhibits food intake
Gall bladder relaxation
Y4 receptors
–
CCK
I cells of duodenum, jejunum; CNS Enteric nerve ending
Food ingestion, protein, fat
Stimulates gall bladder contraction
Stimulates pancreatic exocrine secretion
Delays gastric emptying
Inhibits gastric acid secretion
Reduces food intake
Increases satiety
Stimulates bowel motility
CCK A
CCK B
Multiple
Intestinal
CCK-33, CCK-8
Orexigenic
Ghrelin
Stomach small bowel colon
Hypothalamus
Fasting
Promote GH release
Increases food intake
Promotes gastric motility
Promotes PP release
GHS receptor
–
This chapter discusses the pathophysiological roles of orexigenic and anorexigenic gut hormones in the regulation of food intake.
The Hypothalamic Circuitry
The hypothalamus contains several nuclei involved in appetite regulation including the lateral nuclei, ventromedial (VMN), dorsomedial (DMN), paraventricular (PVN), perifornical and arcuate nuclei (ARC). Anatomically it is in close proximity to the brainstem, amygdala and higher brain centres that are involved in appetite control.
The ARC, positioned at the base of the hypothalamus with an “incomplete” blood–brain barrier, is directly exposed to factors in the systemic circulation such as gut hormones. It contains two subsets of neurons: orexigenic neurons containing neuropeptide Y (NPY) and agouti-related peptide (AgRP), and anorexigenic neurons containing pro-opiomelanocortin (POMC) (Fig. 3.1). The latter is a precursor of α-melanocyte-stimulating hormone (α-MSH), and cocaine- and amphetamine-regulated transcript (CART). α-MSH produces its anorexigenic effects mainly via melanocortin receptors (MCR). Of the five identified MCRs, MC3 and MC4 receptors, present in high density in the PVN, have been shown to be important in the regulation of food intake [1]. α-MSH is a natural endogenous agonist, while AgRP is an endogenous antagonist of the MC3/MC4 receptors [2]. Disruption of the MCR pathway has been shown to be associated with extreme obesity [3]. In fact, transgenic animals lacking the POMC gene, having MC4 receptor mutations or overexpressing AgRP, have been shown to be hyperphagic and obese [3, 4]. In humans, MC4 receptor mutations have been shown to be the most common known cause of single gene (monogenic) obesity [5, 6]. On the other hand, NPY with a shorter half-life compared to AgRP is proposed to exert its effect via Y receptors, subtypes of which have been described (Y1–Y6) (discussed below) [7–10]. NPY has a higher affinity to Y1 and Y5 receptors. These are available in high abundance in the NTS from which NPY neurons project to the PVN.
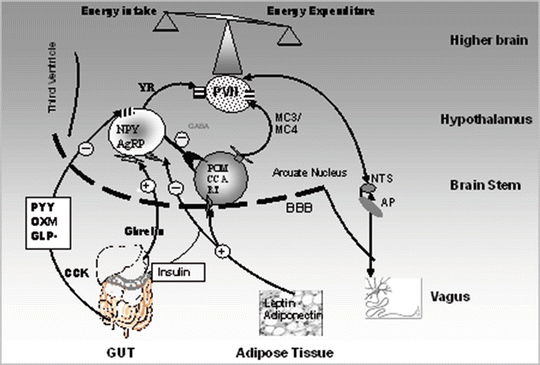
Fig. 3.1
Gut peptide regulation of appetite. Schematic diagram of the gut–brain, gut–gut and gut–adipose tissue interactions. MC3/MC4 R melanocortin 3 and 4 receptors, YR Y receptors, NPY/AgRP neuropeptide Y and agouti-related peptide neurones in the arcuate nucleus in the hypothalamus, POMC/CART pro-opiomelanocortin and cocaine-amphetamine regulated peptide, PVN paraventricular nuclei, NTS nucleus tractus solitarius, AP area postrema, BBB blood–brain barrier
Neuron projections from the ARC extend to the PVN and DMN (Fig. 3.1). Activated neuronal pathways in the PVN signal to the nucleus tractus solitarius (NTS), which also integrates signals from the sympathetic and vagal afferent fibres. The PVN also coordinates input from melanin-concentrating hormone (MCH) producing neurons in the lateral hypothalamus and other brain areas such as the area postrema (AP), regions of the brainstem, and the amygdala that impact food intake. The latter has some areas, which increase feeding and other areas, which inhibit feeding. It is worth noting that areas in the brainstem and amygdala control the mechanics of feeding including salivation, liking, chewing and swallowing. Destruction of these areas would thus cause the animal to lose its recognition for the type and/or quality of food.
The dorsal vagal complex (DVC) comprises the dorsal motor nucleus of vagus (DVN), the AP and the NTS. The DVC is an important part of the brainstem, which transfers peripheral signals from the gut to the hypothalamus via afferent vagal nerves. Mechanoreceptors and chemoreceptors in the GIT activate vagal afferent nerves and these assemble in the NTS. Ultimately, neuronal signals in the NTS conduct signals to hypothalamus. Ascending and descending neuronal projections between the brainstem and hypothalamus are important in the control of food intake [11].
Insulin and glucagon from the pancreas, leptin from adipose tissue and gut hormones (PYY3-36, GLP-1, CCK, and OXM) from the GIT are known anorexigenic hormones. They directly inhibit NPY/AgRP secreting neurons and stimulate POMC/CART neurons in the ARC [12]. Reciprocal to this is the orexigenic gut hormone ghrelin. Ghrelin stimulates NPY/AgRP neurons and inhibits POMC/CART neurons, thereby promoting meal initiation and food intake. While leptin and insulin are known to be long-term regulators of adiposity and energy expenditure, gut hormones are short-lived signals controlling food intake.
Anorexigenic Gut Peptides
PP-Fold Family of Peptides
The PP-fold family of peptides include PYY and PP from the gut and NPY from the central nervous system. These peptides are structurally similar being 36 amino acid peptides containing several tyrosine residues. They are characterised by a specific tertiary structure known as the PP-fold and they become biologically active following COOH-terminal amidation. PP-fold peptides appear to exert their effects through the Y receptors (Y1, 2, 4 and 5) which are classified according to their affinity to PYY, PP and NPY fragments and analogues [7] (Table 3.2). They are all seven transmembrane domain receptors that inhibit adenylate cyclase by coupling to G proteins. The Y1 receptor also increases intracellular calcium and the Y2 receptor regulates calcium and potassium channels. Y1–Y5 are present centrally in the brain, and peripherally in the intestine, pancreas, heart, muscle and blood vessels. They have diverse functions including stimulations/suppression of appetite, reduced intestinal secretion, vasoconstriction and analgesia. Table 3.1 summarises the distribution and functions of these receptors.
Table 3.2
The affinity, distribution and actions of the known Y receptor superfamily
Receptor | High affinity peptide | Low affinity peptide | Distribution | Proposed actions |
|---|---|---|---|---|
Y1 | NPY, PYY | PP | Cortex DRG Amygdala Hypothalamus Blood vessels | Analgesia Anxiolysis ↑ appetite Vasoconstriction |
Y2 (Pre-synaptic) | NPY, PYY, PYY (3-36) | PP | Hypothalamus DRG Hippocampus Intestine | Anorexia Analgesia ↑ Memory ↓ secretion |
Y4 | PP,NPY (2-36), NPY (3-36), PYY, NPY/PYY | NPY/PYY fragments PYY (3-36) | Hypothalamus Amygdala Thalamus Intestine, pancreas, heart, muscle | ↑ Appetite |
Y5 | NPY, PYY, NPY (2-36), NPY (3-36), PYY (3-36), NPY/PYY | NPY/PYY fragments | Hypothalamus Thalamus NTS | ↑ appetite ↑ ACTH |
Y6 (mouse) | PP, NPY/PYY | C-terminal NPY fragments | Intestine Spleen | |
Y6 | A truncated non-functional receptor in man produced by deletion in sixth TM domain | Heart, muscle, intestine, spleen | ||
PYY
PYY was first isolated from porcine intestine [13]. It is produced by entero-endocrine L cells throughout the intestine, but predominantly in the ileum and colon [14, 15]. PYY is co-localised and co-secreted with GLP-1 in response to nutritional stimuli. PYY is released into the circulation 15 min following food ingestion. Once in the circulation, the native PYY1-36 undergoes N-terminal truncation by the action of dipeptidyl peptidase IV (DPPIV) to produce the 34 amino acid form, PYY3-36 [16]. This is the active circulating form of PYY. Post-prandial plasma levels of PYY3-36 plateau after 1–2 h, but remain elevated for up to 6 h.
PYY3-36 is released into the circulation in proportion to the number of calories ingested [17]. Meal composition has also been shown to affect PYY release. Higher plasma levels of PYY are achieved after isocaloric meals of fat compared with that of proteins and carbohydrates [18, 19]. Recent studies in rodents and in man revealed that protein had the most influential effect on PYY release [20], followed by fat and then carbohydrate [21]. Short chain fatty acid (SCFA) infusion into the colon also stimulate PYY release [22] through GPR43 and GPR41 (SCFA receptors) co-expressed on the L-cell apical surface [23, 24]. A number of other factors have been shown to stimulate PYY release including CCK, gastric acid, bile acids, insulin like growth factor-1 (IGF-1), bombesin and calcitonin gene-related peptide (CGRP). Neural signals, such as vagal stimuli, have also been implicated [25, 26]. Evidence to support the latter is the increase in PYY levels in response to the presence of food in the duodenum, before its arrival to the L cells in the ileum. In contrast, release of the peptide is inhibited during fasting [9, 27], and by GLP-1 [28].
PYY Actions
PYY delays gastric emptying and reduces gastric acid and pancreatic secretions [29]. This inhibitory effect is mediated by the stimulation of Y1, Y2 and Y4 receptors on enterocytes and neurons [30, 31]. Infusion of PYY3-36 to healthy volunteers reduces the gut transit [22] probably by its effect on PYY binding sites in the DVC. PYY also affects central appetite regulation [29] and enhances energy expenditure [32, 33].
The peripheral administration of PYY reduces appetite in mice and in man [8, 27]. In mice, administration of PYY3-36 peripherally was shown to acutely reduce food intake. This reduction in food intake continued on chronic peripheral administration of the peptide resulting in reduced weight gain [8]. PYY3-36 has also been shown to inhibit food intake in man. A single infusion of PYY3-36 causes a 30 % and 31 % reduction in food intake in a free-choice meal 2 h post infusion [8, 27] in both obese and lean individuals. Subjective hunger ratings were also reduced with the reduction in calorie intake without changes in gastric emptying [27]. The appetite reducing effect of PYY3-36 persisted for 24 h in both lean and obese subjects, despite PYY3-36 levels retuning to basal levels, which implies that PYY3-36 may be an important physiological post-prandial satiety signal. This supports the findings in the animal studies and suggests that, unlike leptin, PYY resistance [9, 18] seems unlikely.
Insulin sensitivity has been found to be improved by PYY3-36. In animal models of diabetes, long-term peripheral administration of PYY3-36 has been shown to improve glycaemic control as a consequence of reduced food intake, body weight and visceral fat [34].
Mechanism of Action
PYY1-36 binds to Y1, Y2 and Y5 receptors; however, PYY3-36 binds selectively to Y2 receptors. Y2Rs are mainly located in the CNS [35, 36]. The anorexigenic effect of PYY3-36 is absent in Y2 receptor knockout mice [8, 9] and was shown to be completely blocked by Y2 receptor antagonist [37, 38]. Peripheral administration of PYY3-36 has been shown to cause c-fos activation in the ARC [8] demonstrating the ability of peripheral PYY3-36 to activate neurons in this hypothalamic nucleus.
NPY neurons inhibit POMC neurons via GABA mediation. Therefore, inhibition of the NPY neurons results in a reciprocal activation of POMC neurons and induces appetite suppression [39]. However, the anorectic effects of peripheral PYY3-36 were retained in POMC knockout mice raising questions about the importance of melanocortin peptides for the action of PYY3-36. In support of this, MC4R knockout and agouti mice are shown to be sensitive to the anorectic effects of peripherally administered PYY3-36 [40]. These findings support the notion that PYY may exert its effect through multiple pathways. Therefore, it is tempting to propose that although Y1 and Y5 receptors have lower affinities for PYY, they might override the actions of Y2 receptors when they are exposed directly to increasing amounts of PYY. This might be the reason for the contrasting actions of PYY when injected to different parts of the brain. The reduction in the orexigenic effects of centrally administered PYY in both Y1 and Y5 receptor knockout mice [41] further supports this hypothesis. In addition, the AP appears to be yet another brain region through which PYY3-36 exerts its effect. In rats, ablation of the AP results in an increase in the acute anorectic effects of PYY3-36 [42].
PYY Levels in Normal Physiology and Disease
Obese people have relatively lower basal levels of PYY3-36 and have an attenuated surge in PYY3-36 following a meal. This might explain their impaired satiety and greater food intake [43]. They also have lower fasting PYY levels compared with lean subjects [44]. PYY levels are subject to diurnal variation. Levels are higher during sleep (in non-shift workers). Levels are also elevated in cachetic conditions such as cardiac cachexia [45], chronic kidney disease (CKD) [46] and hepatic cirrhosis. In patients with diabetic gastroparesis, there are an increased number of colon cells expressing PYY. This may be the cause of the reduction in gastric emptying and the abnormal gut transit time [47]. PYY may also mediate weight loss following gastric bypass surgery. In wild-type mice, weight reduction following bariatric surgery is associated with increased PYY expression and fasting PYY levels [48]. In contrast, bariatric surgery in PYY KO mice was not associated with weight loss acutely.
PP
PP is produced by PP cells (F cells) within the pancreatic islets [49–51]. It is also expressed sporadically throughout the GIT [30]. Circulating PP levels are subject to diurnal variation being lowest in the early hours of the morning and highest in the evening. Its circulatory half-life is seven minutes [52]. PP is released in response to meal ingestion in proportion to size and caloric content of the meal. The release of the peptide is biphasic, though increasing with consecutive meals [53]. Once in the circulation PP levels remain elevated up to 6 h.
Circulating levels of PP are increased by adrenergic stimulation, ghrelin, motilin and secretin [54–56], and are reduced by somatostatin [57]. Vagal tone also appears to regulate PP release both post-prandially and throughout the day. Propantheline (an anti-muscarinic agent) has been shown to block both the diurnal and the post-prandial levels of PP by 60 %. The latter is shown to be abolished by vagotomy [47].
PP has an inverse relationship with body mass index (BMI) with higher levels in anorectic compared to obese subjects [58, 59]. Transgenic mice, with overexpression of PP, have reduced food intake and lower lean body mass [49]. However, obese animal models show lower sensitivity to the effects of PP compared with the high sensitivity observed in lean animals. Peripheral administration of PP reduces food intake, gastric emptying and also increases energy expenditure through the vagus nerve in mice [51, 60, 61]. In patients with Prader-Willi syndrome, intravenous infusion of PP was shown to reduce food intake [62]. Infusion of PP for 90 min to healthy volunteers reduces food intake acutely and also reduces food intake by 25 % 24 h following infusion [63, 64].
It is thought that PP exerts its anorectic effects via the ARC. PP also signals through Y4 receptors in the vagus nerve [51, 65]. Peripheral PP administration leads to c-fos expression in the brainstem, amygdala and hypothalamus [66]. Manganese-enhanced magnetic resonance imaging (MEMRI) in fasted mice following peripheral administration of PP demonstrates reduced signal intensity in the ARC, VMH and PVN, which correlates with reduced food intake [67]. Other postulated mechanisms for the anorectic effects of PP are via the NPY and orexin pathways and through suppression of ghrelin secretion and vagal neurons. Both NPY protein and mRNA expression are reduced following peripheral PP administration and PP has reduced effects following vagotomy.
PP agonists would be attractive as potential agents for obesity treatment. In 2011, PP 1420, an analogue of PP was developed and used in a phase 1 trial study to assess its tolerability; the results were encouraging and further trials are currently being undertaken [68].
Cholecystokinin (CCK)
CCK was the first hormone recognised to be involved in appetite regulation [69]. It is produced primarily by the I cells in the duodenum and jejunum and to a lesser extent in the ilial mucosa. It is also produced in the brain and by enteric nerve endings where it acts as a neurotransmitter [70]. There are various types of CCK with different lengths. CCK-58, CCK-39, CCK-33 and CCK-8 have all been found in man [71]. The biologically active form of CCK shares a sequence (carboxy-terminus) homology with gastrin [70]. CCK is released post-prandially following exposure of I cells to free long chain fatty acids and amino acids [72, 73]. The cellular pathway, which leads to CCK production, has become clearer in recent years. I cells express GPR40 (a G-protein-coupled receptor), which induces CCK release in response to long-chain fatty acids [74]. It is also recognised that amino acids have a direct effect on I cells by activating calcium sensing receptors (CaSR), which will stimulate CCK secretion [75].
CCK is a known satiety peptide; it slows gastric emptying and inhibits gastric acid secretion, but stimulates intestinal motility, gall bladder contraction, and increases pancreatic exocrine secretion. CCK is known to inhibit food intake in man and in rodents [76]. However the duration of its action is short, with a half-life of only 1–2 min. Therefore, no anorectic effect is observed if CCK is administered more than 15 min before meal intake [77]. Additionally, chronic administration of CCK reduces food intake and increases meal frequency. Consequently, long-term administration does not appear to have any effects on body weight [78]. This suggests that CCK is a short-term inhibitor of food intake.
Mechanism of Action
CCK-1R and CCK-2R belong to the class 1 G-protein-coupled family. CCK activates phospholipase C following binding to these receptors, and this leads to intracellular calcium release [79]. Structurally, there is 48 % similarity in sequence between these receptors [80]. CCK-1R identifies the N-terminal heptapeptide and CCK-2R recognises the N-terminal tetrapeptide, which is similar in CCK and gastrin. This results in greater affinity of CCK-1R to CCK compared to gastrin, whereas CCK-2R has identical binding affinities to both peptides [81]. CCK-1R is the main receptor which modulates food intake and satiety. There are two forms of CCK-1R: high affinity/low capacity and low affinity/high capacity [82]. In mice activation of low and high affinity CCK-1Rs are required to induce satiety, however in rats only the low-affinity CCK-1R activation appears to be important to cause satiety [83]. Pancreas, gall bladder, stomach, kidney, lung and vagus nerve are the main sites that express CCK-1Rs [84]. CCK-1Rs are also present in the brainstem, the hypothalamus; the SON, PVN and DMN, substania nigra, ventral tegmental area and nucleus accumbens [85–87]. There is a high concentration of CCK-2Rs in CNS, however they have a limited peripheral expression in stomach and uterus [84].
Selective CCK-1R agonists cause a reduction of food intake [88]. Although CCK-2R knockout mice are hyperphagic and 28 % heavier than wild-type mice [89], activation and deactivation of this receptor has not revealed any alteration in food intake [90, 91].
The exact role of CCK in inhibition of food intake via the vagus nerve is still unclear. It has been shown that CCK can change gene expression within vagal neurons. Vagal expression of the cannabinoid receptor CB1, melanin-concentrating hormone (MCH) and its receptor MCHR-1 is abolished by activation of CCK-1Rs [92, 93]. It is also recognised that CCK enhances the expression of the Y2 receptor [94], and CART [95] on vagal afferent neurons.
Peripheral administration of CCK, at doses sufficient to inhibit food intake, has been shown to induce synthesis of c-fos in the brainstem, NTS and the dorsal vagal nucleus [96]. Vagotomy blocks the effect of CCK on food intake indicating neuronal requirement for the mediation of CCK action to the CNS [97]. In obesity, because of the decreased electrical excitability, vagal afferent neurons display resistance to the effect of CCK [98].
The Proglucagon Gene Products
Proglucagon is a prohormone containing 160 amino acids with a 20-amino acid signal sequence at the N-terminal end [99]. The preproglucagon gene is expressed in alpha cells within the pancreas, entero-endocrine L cells within the intestine and the NTS in the brainstem [100]. Prohormone convertases 1/3 and 2 convert proglucagon to a number of biologically active fragments. Specifically, in the CNS and L cells OXM and GLP-1 and GLP-2 are produced [101] and in the pancreas, glucagon is produced [102] (Fig. 3.2).
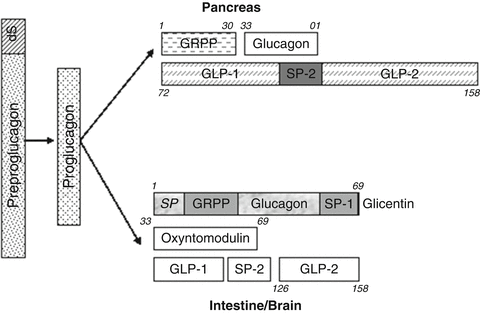
Fig. 3.2
Preproglucagon products. GRPP glicentin-related pancreatic polypeptide, GLP-1 glucagon-like peptide-1 (7-36), GLP-2 glucagon-Like peptide-2, SP-1 spacer peptide-1, SP-2 spacer peptide-2, S signal peptide
Oxyntomodulin (OXM)
OXM is a 37-amino acid peptide produced in the L cells of the intestine along with GLP-1 and GLP-2 [103, 104]. OXM shows diurnal variation with low levels early in the morning and higher levels in the evening [105]. It is released in proportion to food ingestion and calorie intake [103]. Increased plasma levels of OXM have been shown to inhibit gastric acid secretion and motility in both humans and rodents. OXM also stimulates intestinal glucose uptake and decreases pancreatic enzyme secretion in rats. It also causes insulin release via either direct stimulation of β-cell GLP-1R and glucagon receptors (GCGR) or activation of GLP-1Rs on sensory nerves [106, 107].
Administration of either OXM or OXM analogues results in weight loss in obese rats, mice and humans by inhibition of food intake and increases in energy expenditure [108–110].
No specific OXM receptor has been identified. However, OXM is an agonist for both the GLP-1R and the GCGR [101, 111, 112]. OXM reduces food intake by activating GLP-1Rs [113, 114]. Recent studies suggest that OXM exerts its stimulatory effect on energy expenditure via GCGR activation [115, 116] and its glucoregulatory action mostly via GLP-1Rs [117].
Exendin9-39, which acts as a GLP-1 antagonist, can block the actions of both GLP-1 and OXM [18]. GLP-1Rs are present in the NTS and the ARC in addition to its widespread presence peripherally in the GIT, lung, pancreas and heart. Interestingly, exendin9-39 administration into the ARC abolishes the peripheral effects of OXM but not those of GLP-1 [114]. This suggests an ARC site of action for OXM, while GLP-1 acts via the brainstem. Further evidence suggests different neuronal activation patterns between OXM and GLP-1. OXM has a lower affinity (twofold) to GLP-1 receptors compared to GLP-1 [118]. Activation of the neuronal c-fos expression in the ARC, but not in the brainstem region, was observed following intraperitoneal (IP) administration of OXM and exendin9-39 [108, 112, 119, 120]. This pattern of activation is different from that seen following GLP-1 administration [114].
An OXM analogue, selective for GLP-1Rs, has a 100-fold lower effect on liver glycogenolysis. Chronic administration of this analogue to obese mice revealed that weight loss, lipid lowering and anti-hyperglycaemic effects were reduced compared to native OXM. These data support the potential role of GCGR activation by OXM in inducing weight loss [115].
OXM simultaneously activates GCGRs and GLP-1Rs and these have contradictory effects on glucose homeostasis. However, the overall effect is that OXM ameliorates glucose tolerance [117, 121]. In rodents studied with a pancreatic clamp, intrahypothalamic glucagon inhibits liver glycogenolysis and consequently opposes the effects of circulating glucagon to increase liver glucose production. This suggests that activation of GCGR in the CNS following OXM administration in animals may improve glucose metabolism [122].
An additional mechanism whereby OXM may exert its effect on appetite is via suppression of ghrelin. In rodents and humans, peripheral administration of OXM results in a reduction of circulating ghrelin levels by 20 % [114] and 44 % [120] respectively. Human studies on the effects of OXM on appetite control appear to be promising and indicate a novel potential role that OXM or OXM agonists may have as anti-obesity therapeutic agents [108].
Glucagon-Like Peptide-1 (GLP-1)
GLP-1 is secreted from the L cells in response to food intake [123, 124]. Two biologically active potent forms (GLP-17-37




Related posts:



Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
