, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
6.1 Foreign Body Granulomas
6.1.1 Clinical Features
A foreign body granuloma forms as a response of the skin to foreign material in the tissue. Many types of exogenous materials (e.g., glass, metals, and organic materials) and endogenous materials (e.g., hair, keratin, and urates) may trigger the formation of granulomas [1]. Any age or gender may be affected. There is usually a history of a penetrating trauma (frequently to the foot or hand) followed by the appearance of one or more tender papules or nodules in the affected area. If the lesion is on the foot, there may be pain with weight-bearing activity. Symptoms typically develop weeks to years after the inciting event. Treatment is surgical excision.
6.1.2 Histology
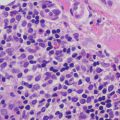
The allergic type of foreign body granulomas contains lymphocytes and epithelioid histiocytes. Multinucleated giant cells are relatively less common. Epithelioid histiocytes aggregate into clusters, forming granulomas. Neutrophilic debris and necrosis are present in some cases.
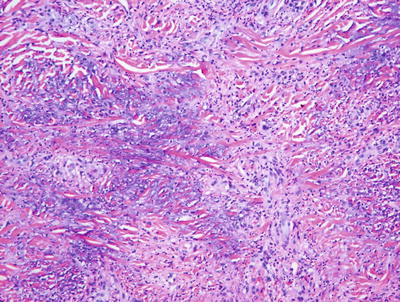
Nonallergic foreign body granulomas demonstrate an admixture of lymphocytes, histiocytes, multinucleated giant cells and, in early lesions, clusters of neutrophils [2, 3] (Fig. 6.1). The most common foreign material that elicits this type of reaction within the skin is keratin , often secondary to a ruptured folliculitis. The keratin is often readily identified within multinucleated giant cells. Other materials may be similarly located and can be visualized with routine microscopy or in some cases highlighted with light polarization (Fig. 6.2). Foci of necrosis are often seen centrally and surrounded by the granulomatous reaction. Changes within the overlying epidermis are related to route of entry of the foreign material into the dermis , and may display ulceration or epidermal acanthosis if the material has been present for a period of time and caused secondary local trauma from rubbing.
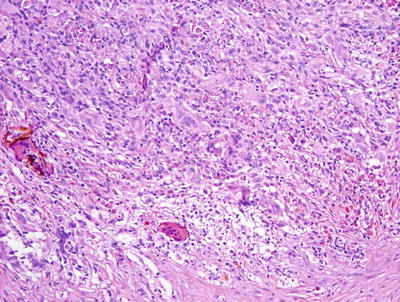
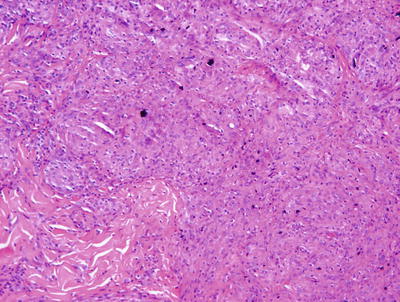
Fig. 6.1
A dense granulomatous infiltrate characterized by multinucleated giant cells and a mixed inflammatory infiltrate is seen in foreign body type granulomas. Foreign material can be apparent on routine H&E sections
Fig. 6.2
In this case of a granulomatous response to a tattoo , abundant dark tattoo pigment is seen both within giant cells and extracellularly
6.1.3 Pathogenesis
Tumor necrosis factor-α (TNF-α) plays a crucial role in inflammatory granuloma formation. TNF-α has been shown to up-regulate adhesion molecules that participate in cellular recruitment and lymphocyte activation. Blocking TNF-α with soluble TNF receptor effectively diminished schistosome-associated granuloma formation and lymphocyte activation in vivo [4, 5]. TNF blockage also reduced intercellular adhesion molecule-1 (ICAM-1) expression. In preclinical animal models , passive immunization of mice with anti-ICAM-1 antibody during primary granuloma formation resulted in an attenuation of lesion development [6].
6.2 Granuloma Gluteale Infantum
6.2.1 Clinical Features
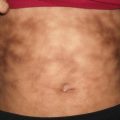
Granuloma gluteale infantum is a rare diaper dermatitis characterized by large, firm, reddish purple nodules or plaques appearing on the inner thighs, perineum, scrotum, and inguinal folds (Fig. 6.3). Granuloma gluteale infantum most commonly affects infants 2–9 months old [7]. Typically, infants have a history of irritant contact diaper dermatitis, with or without secondary yeast infection. There is often a history of previous use of potent topical steroids on the area, suggesting that steroids may be a predisposing factor. Most infants recover completely over time.
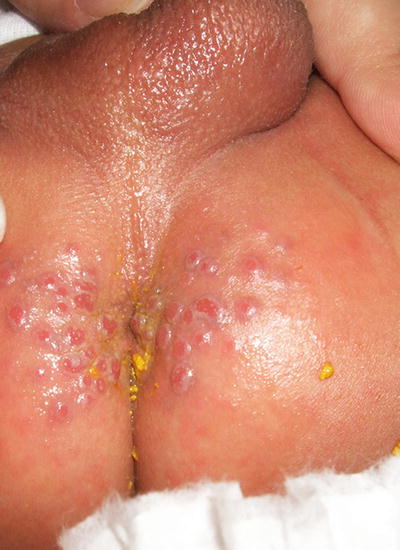
Fig. 6.3
Perianal pseudoverrucous papules and nodules, presenting as eroded papules along the anus and medial buttocks, are thought to be a different presentation of granuloma gluteale infantum
6.2.2 Histology
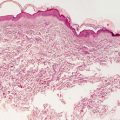
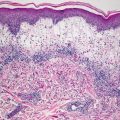
Granuloma gluteale infantum is characterized by hyperplastic epidermis with overlying parakeratosis, indicative of mild chronic external trauma. Within the epidermis , there is a mixed inflammatory infiltrate with neutrophils and eosinophils in addition to lymphocytes and histiocytes (Fig. 6.4). Abscesses containing neutrophils and/or eosinophils may be present within the dermis. Plasma cell may also be seen [8]. In well-developed cases, small granulomas resembling those seen in sarcoidosis are present, but these are not observed in the vast majority of cases [7] (Fig. 6.5). Starch granules have been observed within the granulomatous foci in some cases.
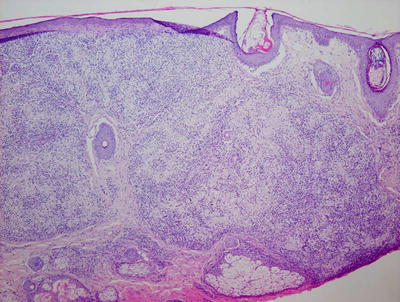
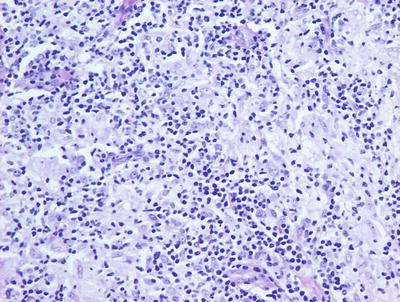
Fig. 6.4
Granuloma gluteale infantum is characterized by a diffuse dermal infiltrate of histiocytes surrounded by a mixed inflammatory infiltrate
Fig. 6.5
Sarcoidal-type granulomas admixed with lymphocytes and scattered neutrophils are observed in well-formed lesions of granuloma gluteale infantum
6.3 Sarcoidosis
6.3.1 Clinical Features
Sarcoidosis is a systemic condition of unknown etiology and is characterized by the formation of non-caseating granulomas in multiple body tissues. Granulomas commonly affect the lungs, eyes, skin, and reticuloendothelial system. Sarcoidosis is most common in young adults. In the pediatric population, it tends to affect adolescents aged 9–15 years old and occasionally preschool-aged children (“juvenile papular sarcoid”) [11]. Initial clinical symptoms are often nonspecific complaints, such as weight loss, fever, fatigue, and anorexia. A variety of pulmonary and eye complaints may occur. Generalized lymphadenopathy is common. About 25 % of patients develop cutaneous involvement at some point during their clinical course [12]. Lesions often appear as red to violaceous or yellow-brown flat-topped papules, nodules, or plaques. There may be an annular configuration. Erythematous, scaly patches and tender subcutaneous nodules are also seen [13]. Ichthyosis, erythroderma, and hypopigmentation have also been reported [14, 15]. Although lesions can occur anywhere, there is a predilection for the face, especially the nares, lips, and eyelids. Lesions may also appear in old scars or other areas of trauma [16].
6.3.2 Histology
Skin biopsies from patients with sarcoidosis may demonstrate various histologic patterns. The primary or specific pattern is a granulomatous dermatitis. The histologic features are those of non-caseating granulomas within the dermis (Fig. 6.6). Well-circumscribed clusters of histiocytes are present at throughout the dermis and in the subcutaneous fat, usually without any changes within the epidermis (Fig. 6.7). The presence of granulomas within the papillary dermis, in close proximity to the epidermis is a relatively specific finding. In most cases, only a scant lymphocytic infiltrate is present around the aggregated histiocytes (giving rise to the “naked granulomas” that are described in this entity). Eosinophils and neutrophils are not abundant in sarcoidosis. Foci of caseation are uncommon in sarcoidosis; however, in some cases, granulomas may be interstitial as opposed to tightly aggregated and may demonstrate focal necrosis [17, 18]. Perivascular and peri-adnexal granulomas can be encountered. Peri-adnexal distribution has been described in hereditary early onset sarcoidosis (Blau syndrome ). Foreign bodies can be identified within granulomas in as many as one-third of cases [19].
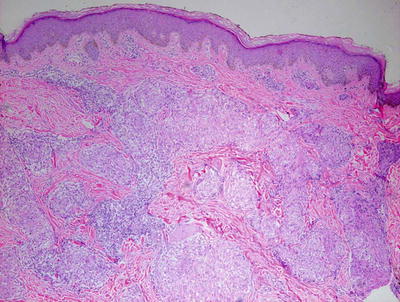
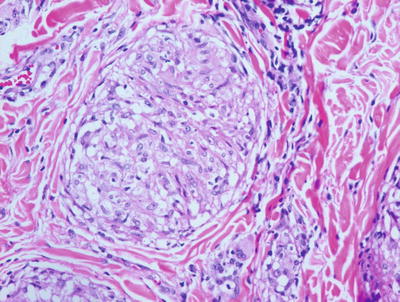
Fig. 6.6
Sarcoidosis demonstrates multiple small well-formed granulomas with a small number of surrounding lymphocytes within the papillary and reticular dermis
Fig. 6.7
Sarcoidosis is characterized by sharply circumscribed granulomas with relatively little surrounding inflammation
Various nonspecific cutaneous eruptions are also commonly encountered in patients with sarcoidosis. The most common histologic pattern seen is erythema nodosum, which is a septal panniculitis that is seen in up to 70 % of these patients [20]. This association is strongest in cases of “acute” sarcoidosis (Lofgren syndrome ) and is more commonly seen in females with the disease. While granulomas may be present within fibrous septa, they are not always seen. Acquired ichthyosis that histologically resembles ichthyosis vulgaris with hyperkeratotic orthokeratosis and loss of the granular layer is also seen in a minority of patients with sarcoidosis [21].
Sarcoidosis should remain a histologic diagnosis of exclusion. While there are characteristic findings, each of these findings can be found in a wide range of other entities, most commonly infectious processes. It is important to perform special stains for microorganisms to exclude infection by fungi and mycobacteria . A search for polarizable foreign material is also recommended before making a diagnosis of sarcoidosis. While not common in children, granulomatous mycosis fungoides should also be considered in cases with a more brisk lymphoid infiltrate.
6.3.3 Pathogenesis
Sarcoidosis has wide variability of disease course and manifestation, thus suggesting that the disease represents a heterogeneous spectrum of disorders. Both genetic susceptibility and environmental factors appear to play a role in disease development. A current concept of the pathogenesis of sarcoidosis is that it is a result of a chronic and aberrant immunological reaction to an antigenic stimulus in genetically susceptible individuals [22–24].
Sarcoidosis has a genetic susceptibility basis that is complex and involves the interplay of multiple genes. The existence of genetic susceptibility is supported by evidence of familial clustering, ethnic variations in disease susceptibility and presentation, as well as the association of this disease with various human leukocyte antigen (HLA) and non-HLA genes [23, 25]. First degree relatives of affected individuals have an increased risk of sarcoidosis as compared to the general population [26]. Several HLA alleles have been found to confer disease susceptibility, including HLA-DR11, 12, 14, 15, 17, HLA-DRB*1101, and HLA-DQB1 [23, 27, 28]. Other HLA alleles found to confer disease protection are HLA-DR1, DR4 and HLA DQ*0202. HLA association suggests that antigen presentation influences sarcoidosis initiation and progression.
Genome-wide association study (GWAS) of single-nucleotide polymorphisms in individuals with sarcoidosis detected a strong association of sarcoidosis with annexin A11 (ANXA11) [29, 30]. ANXA11 association has been found in Europeans, Africans, and European Americans [31, 32]. ANXA11 has regulatory functions in intracellular calcium signaling, cell division, vesicle trafficking, and apoptosis. It has been associated with other autoimmune disorders, including rheumatoid arthritis, lupus erythematosus, and Sjögren syndrome [33]. Another novel risk variant of sarcoidosis has been identified, which is a premature splice site at exon 5 of butyrophilin-like 2 (BTNL2) gene [34].
Infectious pathogens have also been implicated in the etiology of sarcoidosis. Mycobacterium and Priopionibacteria have been suggested to promote granulomatous inflammation in sarcoidosis [35, 36]. However, the role of infectious agents in sarcoidosis is still unclear. Certain environmental and occupational exposures, such as organic dusts, solvents, mold/mildew, pesticides, and wood stoves, have been reported to increase the risk of sarcoidosis [37, 38]. Of note, during the first year following the World Trade Center disaster in 2001, New York City firefighters developed sarcoidosis or “sarcoid-like” granulomatous pulmonary disease (SLGPD) at significantly higher rates than usual [39].
The molecular pathogenesis of granuloma formation in sarcoidosis is characterized by immune responses mediated by type 1 T-helper cells (Th1) and possibly type 17 T-helper cells (Th17), and dysregulated regulatory T-cell function [40]. Cytokines that promote a Th1 response, such as IL-2 and IFN-γ, are upregulated in sarcoidosis and are associated with new-onset sarcoidosis and exacerbation of preexisting disease [41–43].
6.4 Granuloma Annulare
6.4.1 Clinical Features
Granuloma annulare is a fairly common disorder of childhood characterized by grouped papules and nodules in an annular configuration, often over the dorsal wrists, hands, ankles, and feet. At least four clinical variants have been reported: localized, generalized, perforating, and subcutaneous [44]. Granuloma annulare can appear at any age, but it is most common in school-aged children. It is more common in females than males by a 2:1 ratio. Typically, asymptomatic grouped annular papules appear over extensor surfaces. Frequently, lesions are initially misdiagnosed as “tinea corporis ,” but fail to improve with antifungal therapy. In the localized type, skin-colored to pink annular, non-scaly plaques composed of papules and nodules may appear anywhere, but they tend to favor the extensor surfaces of the distal extremities, hands, and feet (Figs. 6.8 and 6.9). Lesions are single or multiple, and typically range in size from a few millimeters to several centimeters. In the generalized form, which is more common in adults, hundreds of small skin-colored to pink papules appear over the entire trunk and extremities. Some may coalesce into small annular plaques. The perforating type is characterized by grouped papules with central umbilication and crusts.
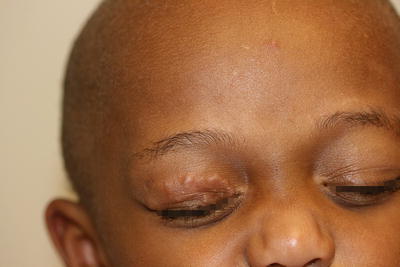
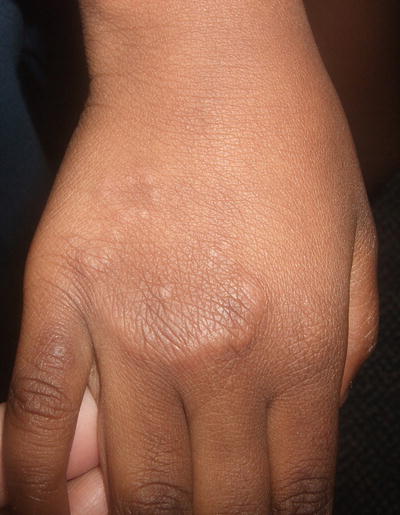
Fig. 6.8
An annular plaque is seen on the eyelid of a child with granuloma annulare
Fig. 6.9
Granuloma annulare presents as papules and nodules forming a non-scaly annular plaque on the dorsal hand
A subcutaneous type of granuloma annulare is relatively common in children, and presents as painless to slightly tender subcutaneous nodules . Although most common on the pretibial surfaces, it has also been reported on other extensor surfaces , as well as on the scalp, eyelids, and buttocks [45–47].
6.4.2 Histology
Pediatric granuloma annulare in many cases differs from that seen in the adult population. While the classic changes of small granulomas occurring within the superficial reticular dermis can be seen in children, the characteristic pattern is that of a single larger palisading granuloma localized in the subcutaneous fat (Figs. 6.10 and 6.11). Less commonly, multiple smaller foci may be present in the deep dermis or subcutaneous fat (Figs. 6.12 and 6.13). This so-called subcutaneous or deep granuloma annulare occurs almost extensively in children. A palisade of histiocytes with abundant eosinophilic cytoplasm surrounds a central area of necrobiosis. In some cases, mucin may be identified within the central foci on either H&E sections or with alcian blue or colloidal iron stains (Fig. 6.14). A surrounding perivascular infiltrate of lymphocytes is present in most cases. Some authors have suggested that the presence of dilated vessels lateral to the granulomatous foci may serve as a diagnostic clue [48]. Classic granuloma annulare demonstrates small granulomatous foci located in the superficial reticular dermis. Palisading histiocytes surround areas of collagen with necrobiosis and mucinous degeneration. Interstitial granuloma annulare also occurs in children. In this variant, the histiocytes percolate individually between collagen bundles and do not form true granulomas. Interstitial mucin deposition is present but subtle (Figs. 6.15 and 6.16).
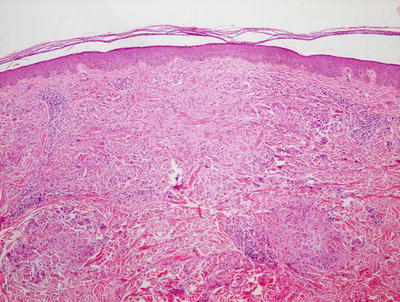
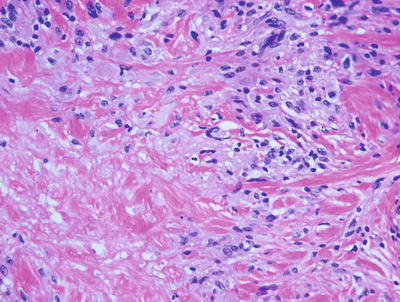
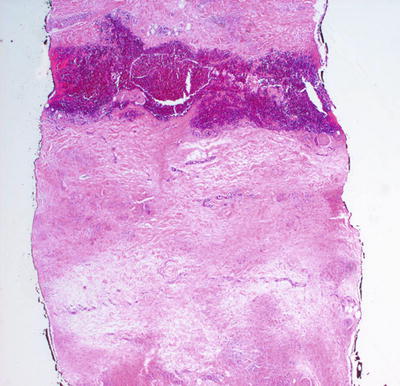
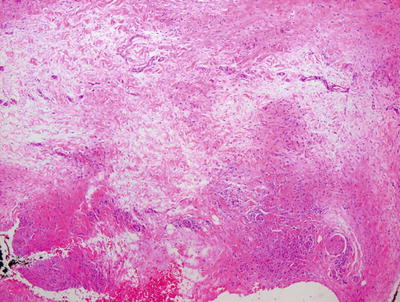
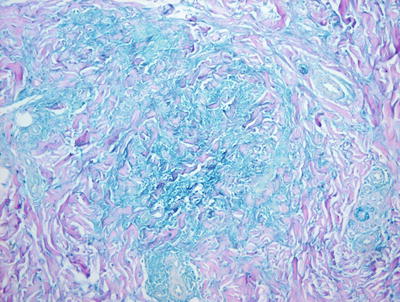
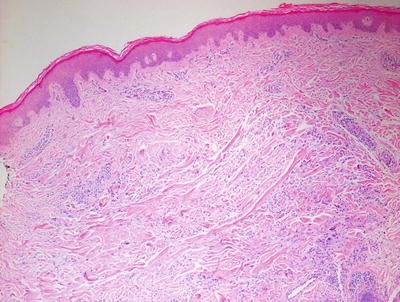
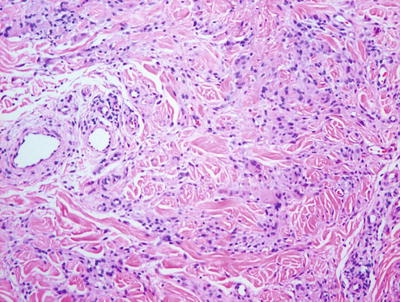
Fig. 6.10
Within the superficial portion of the dermis, several small poorly-formed granulomatous areas are apparent in granuloma annulare
Fig. 6.11
A palisade of epithelioid histiocytes surrounds a paucicellular area of degenerating collagen (necrobiosis) in granuloma annulare. A hint of dermal mucin is present on H&E sections
Fig. 6.12
Similar granulomatous foci are present in the reticular dermis and extending into the subcutis in the deep variant of granuloma annulare
Fig. 6.13
Dermal pallor surrounded by a palisade of histiocytes characterizes the deep variant of granuloma annulare
Fig. 6.14
A colloidal iron stain highlights the blue mucin present within the foci of granulomatous inflammation in granuloma annulare
Fig. 6.15
In the interstitial variant of granuloma annulare , histiocytes percolate individually between collagen bundles in the superficial dermis and do not aggregate into granulomas
Fig. 6.16
Interstitial granuloma annulare shows individual histiocytes percolating between collagen bundles
The major differential diagnoses of granuloma annulare are epithelioid sarcoma and rheumatoid nodule [49]. Epithelioid sarcoma can be differentiated based upon true cellular necrosis and the presence of nuclear atypia in the cells palisading around the periphery of the granuloma-like lesion. In cases that are not easily distinguished on routine sections, immunostains highlighting cytokeratin and CD68 stains will help to make the distinction. A rheumatoid nodule may demonstrate identical histologic features and clinical correlation is the best way to make the distinction.
6.4.3 Pathogenesis
The pathogenesis of granuloma annulare is poorly understood with a vast majority of lesions occurring in patients who are otherwise healthy. Infections, such as tuberculosis, hepatitis and viral infections, have been suggested as a possible cause of granuloma annulare. However, other studies have found no molecular or culture-based evidence for bacterial, mycobacterial, or fungal elements in patients with granuloma annulare [50]. The disease may have a genetic component, since it is associated with certain HLA phenotypes. HLA-B8 levels have been reported to be increased in localized forms of granuloma annulare, and HLA-A31 and HLA-B35 levels are increased in the generalized form [51]. The perivascular lymphocytic infiltrate is mainly composed of T lymphocytes, with a predominance of CD4+ T cells over CD8+ T cells, thus granuloma annulare could be a cell-mediated immune response [52, 53]. Granuloma annulare may represent a delayed-type hypersensitivity reaction, and the matrix degradation (necrobiosis) observed in the disease may be elicited by inflammatory mechanisms.
6.5 Necrobiosis Lipoidica
6.5.1 Clinical Features
Necrobiosis lipoidica is a rare chronic dermatosis that typically affects the lower extremities and is highly associated with type I diabetes. Necrobiosis lipoidica affects 0.3–1.2 % of all diabetic patients (two thirds are with type I diabetes) [54]. In one study, 62 % of patients with necrobiosis lipoidica had diabetes, and nearly half of the patients without clear diabetes had abnormal glucose tolerance tests [57]. The average age of onset is 30 years and it is more common in females [55]. Necrobiosis lipoidica is relatively rare in children with only 2.3 % of reported cases occurring in the pediatric population [56]. Lesions usually begin as erythematous papules that coalesce to form yellowish-brown, telangiectatic plaques with central atrophy and violaceous borders (Fig. 6.17). The classic location is the shins, although lesions have been reported on other body sites, including the arms, trunk, and face/scalp [55]. Ulceration occurs in 35 % of cases [57].
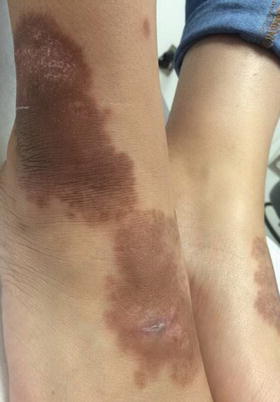
Fig. 6.17
Necrobiosis lipoidica presents as hyperpigmented patches and plaques with areas of atrophy over the dorsal feet and ankles
6.5.2 Histology
Necrobiosis lipoidica is uncommon in children as it is usually a late complication of long-standing diabetes mellitus [58]. However, rare cases have been described in children with long-standing diabetes. In these cases, the histology is indistinguishable from that seen in adult cases of necrobiosis lipodica. Within the reticular dermis, a palisade of histiocytes surrounds a large central area of pale staining necrobiotic collagen (Figs.6.18, 6.19 and 6.21). The histiocytes may have abundant cytoplasm and in well-developed lesions, lipid may be present within the cytoplasm. Multinucleated giant cells may be present. A superficial and deep peri-vascular infiltrate of lymphocytes, histiocytes and plasma cells is present. Similar clusters of hematopoietic cells aggregate into well-formed germinal centers may be present at the interface between the deep dermis and subcutaneous fat (Fig. 6.20). Dermal blood vessels demonstrate thickened walls and in some cases occluded vascular lumen.
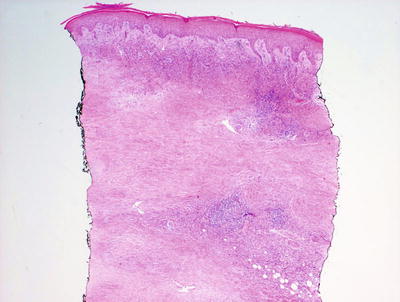
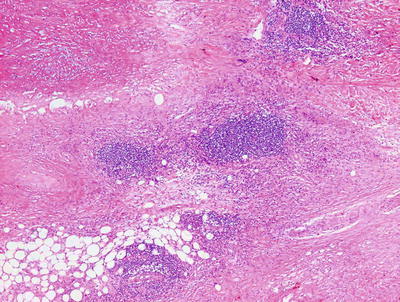
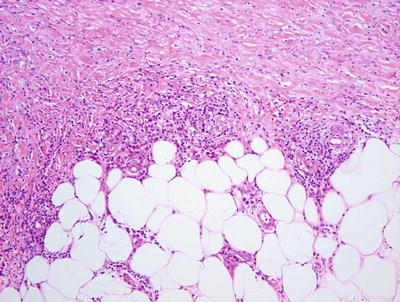
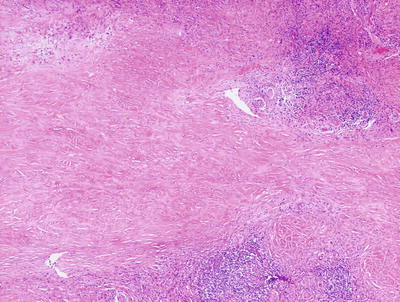
Fig. 6.18
Low magnification demonstrates large foci of necrobiotic collagen surrounded by a rim of histiocytes giving a layered appearance in necrobiosis lipoidica
Fig. 6.19
Necrobiosis lipoidica frequently has lymphoid follicles in addition to the palisade of histiocytes
Fig. 6.20
Lymphoid follicles and plasma cells are often seen at the interface between in the deep reticular dermis and the subcutis in necrobiosis lipoidica
Fig. 6.21
The necrobiotic collagen is lightly eosinophilic and paucicellular in necrobiosis lipoidica
The differential diagnosis includes granuloma annulare, rheumatoid nodule, and necrobiotic xanthogranuloma. The granulomatous foci in granuloma annulare are smaller than those seen in necrobiosis lipoidica and are located either more superficially (classic type) or more deeply (subcutaneous type seen in children). Mucin is not present in necrobiosis lipoidica, unlike the situation in granuloma annulare. Rheumatoid nodules are also situated deeper within the dermis and in the subcutaneous fat than in most cases of necrobiosis lipoidica . In addition, fibrin coats the dermal collagen giving it a “brick red” color in rheumatoid nodule. The distinction is usually best made by the clinical presentation and not based on histologic differentiation. Necrobiotic xanthogranuloma shares many histologic features with necrobiosis lipoidica , but the changes are much more florid. In necrobiotic xanthogranuloma, the dermis is much more extensively necrobiotic with many more histiocytes present that contain cytoplasmic lipids. Multinucleated giant cells are more abundant and much larger in size in necrobiotic xanthogranuloma than in necrobiosis lipoidica.
6.5.3 Pathogenesis
A theory regarding the pathogenesis of necrobiosis lipoidica involves microangiopathy and thickening of the involved blood vessels as a result of glycoprotein deposition in the vasculature [59–61]. Reduced blood perfusion and hypoxia in necrobiosis lipoidica lesions are thought to play a role with increased expression of glucose transporter-1 receptor detected in fibroblasts in the lesions. However, other studies have refuted the microangiopathy theory and suggested that this is a disease that involves an ongoing inflammatory process [62]. Another possible etiology of necrobiosis lipoidica is antibody-mediated vasculitis with involvement of cytokines, complement, and fibrinogen in the disease process [61].
6.6 Rheumatoid Nodule
6.6.1 Clinical Features
Subcutaneous lesions histologically resembling rheumatoid nodules can be seen in children who do not have rheumatoid arthritis. In one group of 15 affected children, the mean age at onset was reported to be 4 years of age [63]. In a second group of 7 affected children, the mean age at onset was 5.5 years of age [64]. Children are usually healthy at the time of lesion onset. Solitary or multiple non-tender, firm, fixed nodules are present on extensor surfaces, such as the pretibial areas, dorsal feet, and fingers. Lesions have also been reported on the scalp. They range in size from a few millimeters up to 5 cm. There is often a history of sudden onset and rapid increase in lesion size. Nodules tend to remit spontaneously over several years [63].
6.6.2 Histology
True rheumatoid nodules are rare in children and histologically indistinguishable from subcutaneous granuloma annulare. One large or several smaller palisades of histiocytes are present around a central area of altered collagen (Figs. 6.22 and 6.23). In some cases, the collagen demonstrates an accentuated red color due to fibrin deposition. In other cases, there is a bluish tinge on account of mucin deposition. Clinical correlation is the only reliable way to make the proper diagnosis. In some patients with juvenile rheumatoid arthritis , the increased number of rheumatoid nodules has been associated with methotrexate treatment [65].
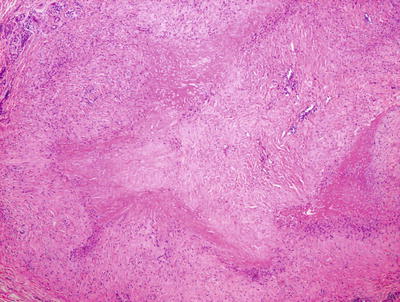
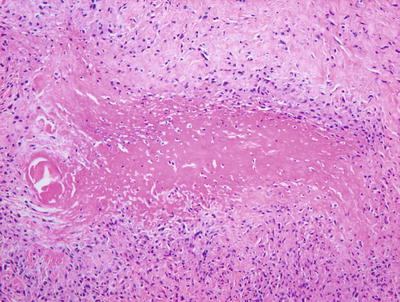
Fig. 6.22
Rheumatoid nodule demonstrates a large focus of eosinophilic collagen with a surrounding palisade of histiocytes in the deep dermis and subcutis
Fig. 6.23
The dense eosinophilia is due to fibrin deposition on the collagen bundles in rheumatoid nodules
6.6.3 Pathogenesis
Rheumatoid nodule is a common skin manifestation of rheumatoid arthritis. The nodule is usually found on pressure points (such as the olecranon process ), but it can occur at any body site. Rheumatoid factor is often present in patients with rheumatoid nodules [66]. Smoking may increase the risk of developing these lesions [67]. Complement activation, immune complex deposition in blood vessel walls, and pro-inflammatory cytokines have been implicated in the pathogenesis of rheumatoid nodule [68].
6.7 Palisaded Neutrophilic and Granulomatous Dermatitis
6.7.1 Clinical Features
Palisaded neutrophilic and granulomatous dermatitis is a rare inflammatory dermatosis most commonly seen in adults with systemic lupus erythematosus, rheumatoid arthritis, and systemic vasculitis (particularly Churg–Strauss syndrome and Wegener’s disease ). It is rare in the pediatric population with only several known case reports in children have been published. Cases in children have occurred in the setting of diabetes, celiac disease, systemic lupus erythematosus, and pauci-immune focal segmental glomerulonephritis [69]. Children with palisaded and neutrophilic dermatitis usually have small erythematous papules and nodules scattered over the extensor surfaces, such as the ankles, feet, elbows, knees, and hands. Papules may coalesce into a linear or annular distribution. Linear subcutaneous bands may also occur [70]. Adults are more likely to present with linear subcutaneous bands on the trunk.
6.7.2 Histology
Palisaded neutrophilic and granulomatous dermatitis is often associated with collagen vascular diseases [71]. The histologic features include leukocytoclastic vasculitis associated with foci of collagenous degeneration in early lesions (Figs. 6.24 and 6.25). As lesions become more fully developed, areas of leukocytoclasis with fibrin deposition and degenerated collagen are found in the centers of palisading histiocytes (Fig. 6.26). Later lesions have fibrosis, minimal neutrophilic debris, and less cellularity.
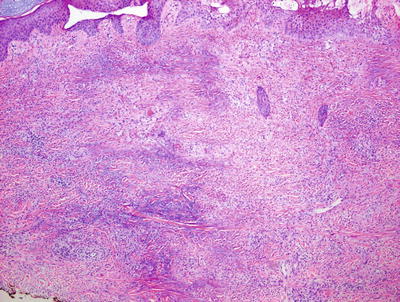
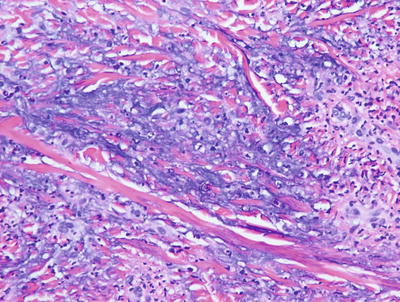
Fig. 6.24
A diffuse infiltrate of histiocytes and neutrophils is seen throughout the dermis in palisaded neutrophilic and granulomatous dermatitis
Fig. 6.25
There is abundant interstitial mucin admixed with neutrophils and karyorrhectic debris in palisaded neutrophilic and granulomatous dermatitis