, Jane Tomimori1, Sofia Beatriz Machado de Mendonça1 and Douglas Antonio Rodrigues1
(1)
Universidade Federal de São Paulo, São Paulo, Brazil
7.1 Epidemolysis Bullosa
Epidermolysis bullosa is a group of genetic diseases characterized mainly by the appearance of vesicles and blisters on the skin and mucous membranes, mainly after minor trauma, due to defects in the composition of the cellular ultrastructure of the skin that results in skin fragility. There are forms with localized lesions and extensive lesions (Figs. 7.1, 7.2, 7.3, and 7.4) . They are classified as simple, junctional, dystrophic, and Kindler syndrome, depending on the structures and location of the basic defect. These major forms are also subdivided into other forms. In the simple forms the formation of blisters is intraepidermal. The clinical presentations are variable and usually do not leave scars, and there is no compromise of the general state. In the juncional forms , the formation of blisters occurs at the level of the lucid lamina. The clinical presentations are variable, with some very severe forms that can lead to death by complications. In the dystrophic forms the formation of blisters occurs just below the dense lamina of the basement membrane of the epidermis. In addition to the formation of blisters and scars, the nails become affected and the formation of milia is observed.
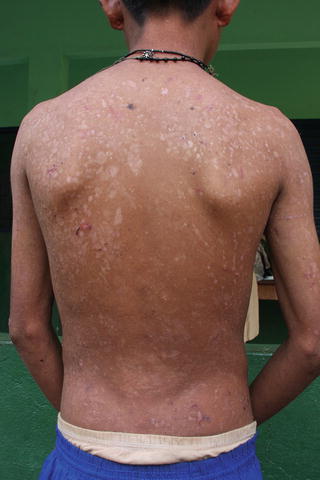
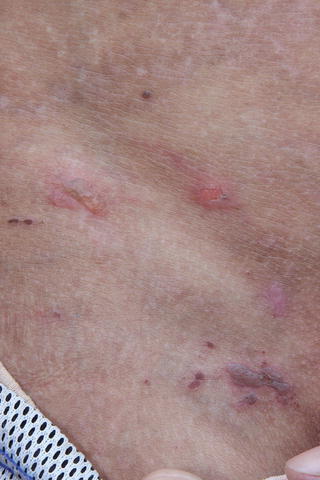

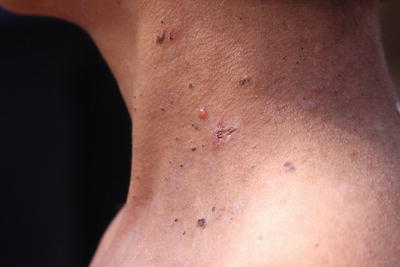
Fig. 7.1
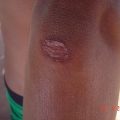
Epidermolysis bullosa: lesions and scars on dorsum
Fig. 7.2
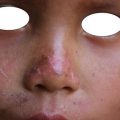
Epidermolysis bullosa: vesicles and blisters
Fig. 7.3
Epidermolysis bullosa: vesicles and blisters on hands
Fig. 7.4
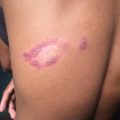
Epidermolysis bullosa: vesicles and blisters on neck
In some dystrophic forms the condition is more severe and extensive and is associated with weight loss, chronic anemia, synechiae in the hands and feet, and unfavorable prognosis. Squamous cell carcinoma may arise in areas with chronic lesions. Laboratory diagnosis can be made through skin biopsy for anatomopathological examination. To define the level of ultrastructural involvement, other tests are necessary (immunomapping, direct immunofluorescence, immunohistochemistry, or electron microscopy).
For differential diagnosis , the possibility of other genetic or autoimmune bullous dermatoses and bullous impetigo must be evaluated. For therapy, it is important to avoid cutaneous trauma, in addition to the application of dressings, dental treatments, dietary regulation, pain and pruritus treatment, prevention of synechiae, and treatment of complications (e.g., infections).
7.2 Peutz-Jeghers Syndrome
Peutz-Jeghers syndrome is an autosomal dominant inherited disorder. It is characterized by the presence of mucocutaneous lentigines associated with gastrointestinal polyposis. The oral mucosa is the site most suitable for physical examination, contributing to the early detection and diagnosis of the disease. The disease is linked to a higher incidence of colorectal carcinoma This disease is of familial occurrence and emerges after birth or during childhood.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


