Fig. 2.1
A simplified overview of research study designs. Observational studies, where no intervention is introduced, include cohort, cross-sectional, and case control studies. In contrast, interventional trials include an intervention of interest and are most commonly designed as randomized control trials (RCTs) which can utilize parallel or crossover treatment and placebo groups
Inset 2.1
Example of a randomized trial:
Clobetasol propionate, 0.05 %, vs hydrocortisone, 1 %, for alopecia areata in children: a randomized clinical trial. Lenane P, Macarthur C, Parkin PC, Krafchik B, DeGroot J, Khambalia A, Pope E. JAMA Dermatol. 2014 Jan;150(1):47–50.
In this single-center trial, a low- and high-dose topical steroid were compared over a 24-week period on children ages 2–6 with alopecia areata of the scalp. The trial was blinded in a 2-arm parallel group. Topical steroids were applied for 6 weeks on and 6 weeks off for two cycles during the 24-week study period. The primary endpoint assessed was hair loss at the end of the study. Investigators noted a greater decrease in hair loss in the high potency group compared to the cortisone group. One subject in the high potency group had atrophy which resolved in 6 weeks. No systemic cortisol disturbances were observed.
In RCTs, two groups of patients are established: those receiving the treatment being studied, and those who do not receive the study treatment, but rather a placebo (or a previously established treatment) [2]. The advantages of this design allow for bias reduction through randomization [5]. Bias occurs when different variables such as age or gender are not balanced between patient groups and therefore sway trial results. Randomization refers to a patient being randomly assigned to either the treatment group or placebo (control) group once they have been screened and identified as being eligible for study participation [11]. Randomization reduces bias by randomly distributing these variables, ideally equally, between the groups [6].
Inset 2.2
Blinding or Masking
In a study of hypnotism, or mesmerism, Benjamin Franklin, and Antoine Lavoisier blindfolded (or masked) subjects to prevent them from seeing treatments before evaluating the claimed results. Though used interchangeably with blinding, masking implies eye openings and the ability to see what is going on. Because of potential confusion, blinding has become the standard term in the international clinical research lexicon.
Franklin B, Bailly JS, Lavoisier A. Rapport des commissaires chargés par le roi, de l’examen du magnetisme animal. Chez Gabriel Floteron: A Nice; 1785.
RCTs can be further modified by their design and degree of blinding. RCTs typically utilize a parallel-group design in which the two groups remain separate in their treatment setup, but everyone within each group is treated identically. Crossover studies are studies in which each patient receives both the study intervention and the control for equal but separate time periods. These also offer the benefit of further bias reduction since each patient serves as his own control [12]. Also critical to the design of a RCT is the blinding status. In a single blind RCT, the investigator is aware of a patient’s treatment status, but the patient is not [11]. This design leaves the data vulnerable to experimenter’s bias in which the investigator’s knowledge of treatment status could influence his evaluation [13]. Double-blind studies eliminate this bias, as well as the placebo effect, because neither the investigator nor the patient is aware of treatment status [11]. An outside participant, typically an unblinded pharmacist, is the one who is aware of the patient’s treatment status. An understanding of how RCT design and blinding allows investigators to develop the RCT that will investigate their hypothesis while limiting the degree of bias involved.
Inset 2.3
Example of a multicenter DBPCR randomized.
Omalizumab for the treatment of chronic idiopathic or spontaneous urticaria.
Maurer M, Rosén K, Hsieh HJ, Saini S, Grattan C, Gimenéz-Arnau A, Agarwal S, Doyle R, Canvin J, Kaplan A, Casale T. N Engl J Med. 2013 Mar 7;368(10):924–35.
This was a phase-three trial to evaluate the safety and efficacy of omalizumab in anti-histamine refractory chronic idiopathic urticaria. Volunteers were randomly assigned to receive drug at three different doses, or placebo, followed by a 4-month observation period. They were asked to score their itching. The baseline itching score was 14 in all four groups. It dropped to 9 in the placebo group, 8, 6, and 4 in the 75 mg, 150 mg, and 300 mg groups, respectively.
2.2 Phases of Drug Development
For a new drug to be approved by the FDA for commercial use, it must undergo a series of trials (Fig. 2.2). The process begins when a new drug is developed in a laboratory setting. Laboratory testing usually begins with cell studies, and ultimately graduates to live animal studies to determine pharmacokinetics and toxicity [14]. For this preclinical testing data to be considered acceptable to the FDA, it must comply with good laboratory practices (GLP). Adhering to GLP ensures that the data produced in the laboratory studies meet the minimum environment, personnel, and technique standards necessary to ensure reliable data. The specific goals that contribute to a successful preclinical trial include understanding basic pharmacokinetics of the drug, identifying drug toxicity levels in two different species of animals, and performing short-term toxicity studies that are approximately equal in time length to the actual drug treatment time [15].
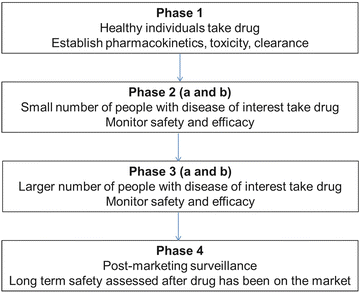
Fig. 2.2
A diagram of study progression. All investigational drugs or products that pass preclinical testing and are reviewed by the FDA are first tested in a phase 1 clinical trial and then progress to phase 2 and 3 trials. Once approved by the FDA, phase 4 studies continue to monitor the product for long-term safety purposes
Once acceptable standards are met in animal models, the drug is then studied in humans. This begins with a Phase 1 trial in which a small number of healthy individuals take the drug. These participants are then studied to further establish pharmacokinetics and toxicity as well as drug clearance in humans. Again, once an acceptable standard is met, the drug passes to a Phase 2 trial where its safety and efficacy are measured in a small number of patients who have the disease the drug intends to treat [14]. Of note, Phase 2 trials can be split into Phase 2a and Phase 2b trials. Phase 2a trials focus on proving the suspected mechanism of action of the drug and typically involve fewer patients than Phase 2b trials. Phase 2b trials strive to identify the ideal dosage of the study drug that allows for the desired efficacy while minimizing side effects [16]. If the drug is shown to be effective for the disease of interest, it is then tested in a Phase 3 trial. Phase 3 trials continue to demonstrate efficacy and safety in patients who suffer from the disease of interest, but involve a much larger patient population and test the drug at different concentrations as well as in combination with other medications [14]. Similarly to Phase 2 trials, Phase 3 trials can be split into Phase 3a trials where the main goal is to generate sufficient data to demonstrate safety and efficacy, and Phase 3b trials which seek to support future publications [16]. Finally, when sufficient data has been collected, the sponsor of the drug submits a new drug application (NDA) to the FDA for final approval. Phase 4 trials, or post-marketing surveillance, are conducted after an approved medication is on the market in order to test long-term safety of the medication [14].
Under the FDA Amendments Act (FDAAA) of 2007, pharmaceutical companies are tasked with maintaining standards of transparency regarding their study data. The main goal of the FDAAA was to ensure that the FDA received the necessary resources to review new trials; however, the act also impacted the degree of transparency of sponsor-initiated clinical trial data. The FDAAA requires “disclosure of any restrictions on public presentation or publication of results of studies funded by industry” [17]. Now drug companies are required to make available to the public, information and results regarding their clinical studies, regardless of the stage of drug development. Most industries list their studies and relevant information regarding the studies and medications on their website so that the public can learn more about the methods, goals, and safety of current research. The public can also learn more about the multitude of clinical studies being performed by visiting www.clinicaltrials.gov.
Inset 2.4
The Sponsor Team
Clinical Research Associate (CRA): The CRA is also called the monitor. Budget time to meet with the monitor and try to make a good impression. A typical trait for a monitor to have is compulsive attention to detail. The frequency and intensity of monitor visits vary with the experience of the investigative site, the complexity of the trial, and the dictates of the protocol. The monitor makes sure that your site is conducting a study according to the protocol, that your data are accurate, complete, contemporaneous, legible, attributable, original, and enduring. The monitor makes sure any deviations from the protocol are adequately explained. The monitor ensures that adverse events are promptly and correctly addressed. The monitor takes your questions and concerns back to the sponsor for feedback. Your monitor is a dedicated, knowledgeable professional, who may have even been a CRC once. Be very courteous, respectful, and attentive to your monitor’s needs. A good relationship with your monitor will make your study go very smoothly. Your monitor may also be in the loop for a variety of studies, and will likely recommend you and your site if you perform well. Pay close attention to any concerns your monitor has. These should be addressed promptly, courteously, and professionally. Your monitor knows the protocol, and has been to a number of sites to see how the protocol is executed. Any lapses or concerns your monitor observes including out-of-date training certificates or unsigned documents or disorganized documents should be taken seriously. By doing so, not only will you improve the quality and timeliness of your work, you will avoid trouble in case of an audit. You will also save money, and make your monitor and sponsor happy. Visits can last from 4 h to several days. Disorganization costs money. Monitor visits to disorganized sites take longer, and often require revisits to ensure accuracy of data. This means more travel costs and more time costs for the sponsor. Your CRA may also be monitoring several sites (typically 5–10). If your site is not organized, you will be costing the monitor time away from family and from other sites.
Medical Research Associate (MRA): The MRA an in-house CRA at the sponsor’s facility. The MRA may oversee CRAs and studies and monitor subject safety, and make sure that all procedures are conducted in accordance with the protocol. The MRA may have been a CRA in the past and may be tapped to cover for your CRA if he or she is on leave or vacation or transitioning to another study. Give the same courteous treatment to your MRA that you do to your CRA.
Sponsor: This is the overall developer of the drug or device. The sponsor oversees the development of a device from its initial chemical identification all the way through manufacturing, testing, approval, marketing, and post-marketing phases. The sponsor finances all aspects of a study from designing the trial, to providing materials, collecting data, monitoring trial, auditing all procedures and data to support the application to the FDA. Sponsors also keep investigators informed about the drug, including new safety information.
Medical monitor: a physician at the facility who is on call for questions about the protocol or safety issues.
Inset 2.6
Contract Research Organization (CRO): As the term implies, this is a groupe hired or contracted out by the Sponsor to administer the trial. Clinical trials are often the most expensive part of investigational research. To successfully usher an investigational product from the clinical phase to the marketing phase can require hundreds of research sites, thousands of study volunteers, and millions or billions of dollars. To save money, and to have a relatively fixed handle on the cost of each phase of a trial, a sponsor may hire a CRO or Academic Clinical Trials Unit (ACTU) to administer a study. A CRO may also have a niche, such as dermatology (e.g., DermTech), and provide resources and expertise to a smaller pharmaceutical company that may not have the staff to dedicate to trial administration. Working with a CRO can provide you with access to a study and help you build your portfolio of clinical research. The drawback to working with a CRO is that administrative fees taken by a CRO amount to a “tax” on your revenue.
Site Management Organization (SMO): An SMO is essentially a CRO, but one that is affiliated with a site, such as a hospital or academic institution. If you are in private practice and work with an SMO, you have to make sure your contract has legal protections for you regarding intellectual property, Anti Kickback Statutes, and Stark Laws. Site management organization, manages a number of sites in its network. SMOs are also proliferating internationally, where costs are less, but where oversight is also more difficult.
Inset 2.5
Large multicenter trials are often complex and involve several sites. They can be expensive to conduct. They are often more time consuming for investigators because there are more documents, and adverse events to review. The trial below has pooled data from a number of sites and has a detailed Supplementary Appendix to satisfy reproducibility requirements.
Four large multicenter randomized double-blinded studies examined the response to placebo or ingenol mebutate gel. The number of actinic keratosis on either the face and scalp or trunk and extremities were assessed during the study. Data were pooled from similar skin sites and compared.
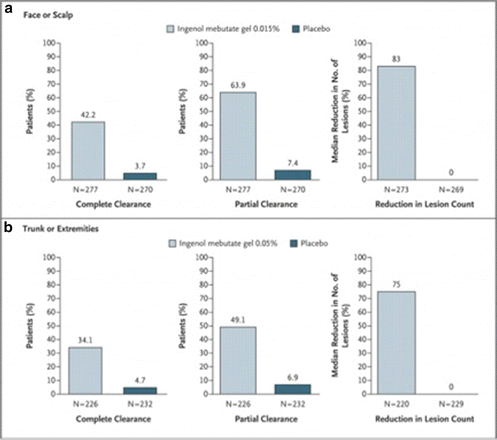
Lebwohl M, Swanson N, Anderson LL, Melgaard A, Xu Z, Berman B. Ingenol mebutate gel for actinic keratosis. N Engl J Med. 2012 Mar 15;366(11):1010–9.
Inset 2.7
A listing of clinical trials can be found at the following web site: www.clinicaltrials.gov. An example of a pilot study on a rare genodermatosis using siRNA is a study of TD101:http://www.clinicaltrials.gov/show/NCT00716014.
This is a first in humans Phase I dose-escalation trial of an interfering RNA in a dominant negative genodermatosis.
Sancy A Leachman, Robyn P Hickerson, Mary E Schwartz, Emily E Bullough, Stephen L Hutcherson, Kenneth M Boucher, C David Hansen, Mark J Eliason, G Susan Srivatsa, Douglas J Kornbrust, Frances JD Smith, WH Irwin McLean, Leonard M Milstone, Roger L Kaspar. First-in-human Mutation-targeted siRNA Phase Ib Trial of an Inherited Skin Disorder. Mol Ther. 2010 February; 18(2): 442–446.
In vitro studies show dominant interference of keratin filament function.
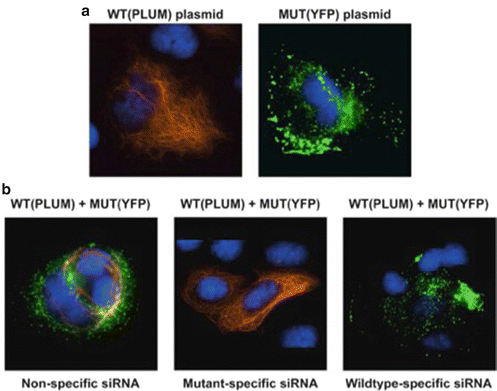
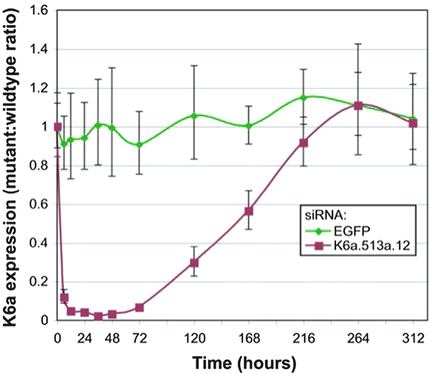
Dose of administered siRNA injected into subject lesions was escalated over 119 days, to a maximal concentration of 8.5 mg/mL and a total dose of 17 mg.
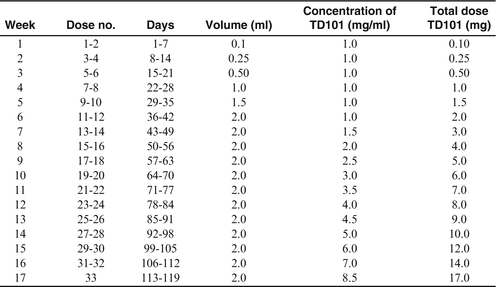
The patient assessed improvement of plantar skin thickness on the vehicle side and treated side.
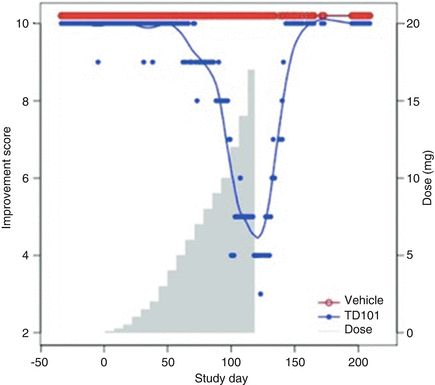
Investigators measured callus thickness on treated and placebo surfaces.
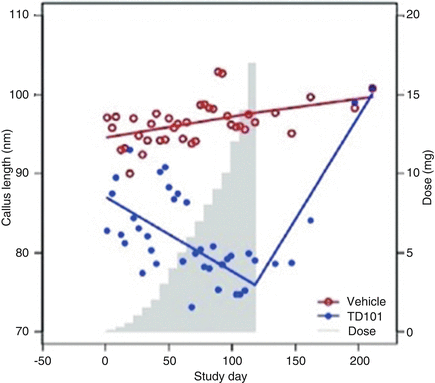
Photographs were taken which prevented identification of the subject.
2.3 Evolution of US Drug Law
Essential to the development of clinical trials are the drug laws that have established an acceptable degree of safety and efficacy for newly manufactured drugs. The first form of organized US drug law was developed in 1820 with the establishment of the US Pharmacopeia (USP), the first official list of standard drugs used in the United States [18]. Over time, the laws have evolved to keep up with advancement in science and engineering (Table 2.1). However, their goals remain the same—to ensure the efficacy of new investigational products as well as the safety of the patients who contribute to their development. Certainly their influence on clinical trials warrants a brief discussion of the history of their evolution and impact.
Table 2.1
Brief timeline of US Drug Law Evolution
1820 | US Pharmacopeia established |
1902 | Biologics Control Act |
1906 | Pure Food and Drugs Act |
1911 | US vs. Johnson |
1912 | Sherley Amendment |
1927 | Bureau of Chemistry Splits |
1930 | Regulatory Branch of Bureau of Chemistry is renamed FDA |
1962 | Kefauver-Harris Drug Amendments Act |
1983 | Orphan Drug Act |
1998 | Pediatric Rule |
2003 | Pediatric Research Equity Act |
Before the direction and organization offered by drug laws existed, drug manufacturers didn’t follow a standard protocol; this led to inconsistencies in drug development and sanitation. The consequences of these practices came to national attention in 1901. At that time, scientists were developing diphtheria vaccines by injecting Corynebacterium diphtheriae into horses and collecting their antibody-rich serum. However, due to a lack of sanitary protocol, thirteen children were killed after they were accidentally exposed to tetanus toxins incurred from this practice [19]. The tragedy led Congress to establish the Biologics Control Act, which was tasked with overseeing the safety and purity of vaccines. Five years later, the Biologics Control Act was molded into the Pure Food and Drugs Act of 1906 by then-president Theodore Roosevelt with the goal of blocking foreign trade of “mislabeled food and drug products” and prosecuting those who were found guilty of these practices [20]. The law also required that each drug should have a label of active ingredients and should maintain a minimum drug purity level set by the US Pharmacopeia [21].
Whereas early US drug laws focused on purity and safety in manufacturing of drugs, more recent drug laws have focused on the importance of data from clinical trials in establishing the potential for adverse events and drug safety. In 1962, a new drug, thalidomide, gained popularity in Europe for its use as a sedative as well as an off-label use as a cure for morning sickness during pregnancy. However, doctors soon discovered that thalidomide was responsible for thousands of infants being born with phocomelia, or dysmorphic limbs [22]. Fortunately, the Food and Drug Administration (FDA) had not given approval for this drug due to FDA inspector Frances Kelsey’s demand for data from clinical trials and for more convincing evidence that the drug did not cross the placenta [23]. The disastrous outcomes from this ordeal led to the development of the Kefauver-Harris Drug Amendments Act of 1962 which increased monitoring of drug approval processes as well as required clinical trial data demonstrating the safety and efficacy of new drugs before drugs could be approved.
After drug laws addressed the need for legitimate data from clinical trials for drug development, they shifted to focus on the different needs of specific patient populations. For example, in 1983, the Orphan Drug Act was passed which allowed the FDA to promote research for drug development for particularly rare diseases since they would otherwise not receive much attention [20]. Since this act passed, orphan drugs have continued to receive increased attention. As an example, the National Institutes of Health (NIH) has created the Therapeutics for Rare and Neglected Diseases (TRND) program that offers incentives for collaborators, including academic scientists, non-profit organizations, and pharmaceutical companies, to apply to work with NIH research teams to promote research efforts for new orphan drugs. The overall goal of these collaborations is to expedite the time necessary for a new drug discovery to progress through preclinical testing so that it may be a suitable project for pharmaceutical companies interested in developing the necessary clinical trials [24, 25]. In 1998, the FDA promoted the Pediatric Rule which extended the mandates of the Kefauver-Harris Drug Amendments Act to drugs that would be applicable to pediatric patients. This, in combination with the Pediatric Research Equity Act of 2003, which grants the FDA authority to mandate that sponsors conduct research for pediatric applications of investigational drugs, ensured that research for new drugs adequately addressed the needs of pediatric patients [20]. Obviously, medical knowledge continues to expand and offer new therapies to different patient populations. Just as important, however, is the fact that drug laws continue to evolve and direct drug development to protect the patients who need them.
2.4 How to Initiate Clinical Trials or Start a Clinical Research Site
There are three main characteristics that Sponsors and contract research organization (CRO’s) look for in a site that is interested in doing clinical trials: the principal investigator (PI) qualifications, site adequacy, and patient population.
2.4.1 PI Qualifications
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


