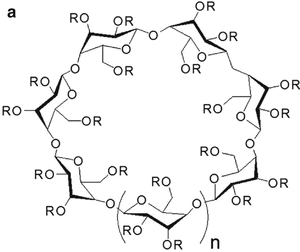
Cyclodextrin | n | R = H or | Abbreviation | Synonyms | MSa | MW (Da) | Solubilityb (mg/ml) | LogK o/w c |
|---|---|---|---|---|---|---|---|---|
α-Cyclodextrin | 0 | αCD | Alfadex | – | 972.8 | 130 | −13 | |
β-Cyclodextrin | 1 | βCD | Betadex | – | 1,135 | 18.4 | −14 | |
2-Hydroxypropyl-β-cyclodextrin | 1 | −CH2CHOHCH3 | HPβCD | Hydroxypropyl betadex | 0.65 | 1,400 | >600 | −11 |
Sulfobutylether β-cyclodextrin sodium | 1 | −(CH2)4SO3 − Na+ | SBEβCD | Betadex sulfobutyl ether sodium | 0.9 | 2,163 | >500 | <−10 |
Randomly methylated β-cyclodextrin | 1 | −CH3 | RMβCD | 1.8 | 1,312 | >500 | −6 | |
γ-Cyclodextrin | 2 | γCD | Gammadex | – | 1,297 | 249 | −17 | |
2-Hydroxypropyl-γ-cyclodextrin | 2 | −CH2CHOHCH3 | HPγCD | Hydroxypropyl gammadex | 0.6 | 1,576 | >500 | −13 |
The regulatory status of cyclodextrins is slowly evolving as more and more cyclodextrin-containing products are being approved (Hincal et al. 2011). All three parent cyclodextrins and many of their derivatives can be found in US Pharmacopeia/National Formulary (USP/NF), the European Pharmacopoeia (Ph.EUR.), and the Japanese Pharmaceutical Codex (JPC). The parent cyclodextrins have been included in the “generally recognized as safe” (GRAS) list of the FDA, and they are commonly found in both food and toiletry products throughout the world. Worldwide cyclodextrins can be found in about 40 marketed pharmaceutical products (Loftsson and Brewster 2010; Hincal et al. 2011).
14.1.2 Cyclodextrin Complexes
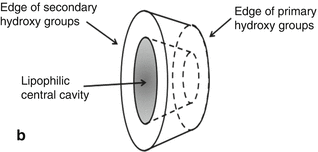
Cyclodextrins are able to form drug-cyclodextrin inclusion complexes by taking up somewhat lipophilic drug moieties (or even small lipophilic molecules) into the central cavity (Fig. 14.1). No covalent bonds are formed or broken during the complex formation, and drug molecules bound in the complex are in very dynamic equilibrium with free drug molecules in solution. Thus, cyclodextrin complexes dissociate readily upon simple dilution, for example, upon injection into liquid chromatographic system or after parenteral administration.
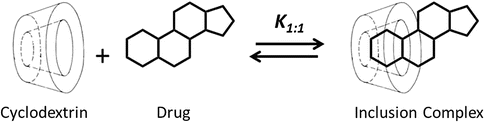
Fig. 14.1
Formation of one-to-one (i.e., 1:1) drug-cyclodextrin inclusion complex
The main purpose for adding cyclodextrins to percutaneous drug formulations is to enhance aqueous solubility of poorly soluble drugs and, thus, increase their topical bioavailability. Higuchi and Connors’ phase-solubility method is used to study the effect of cyclodextrin concentrations on drug solubility (Fig. 14.2) (Higuchi and Connors 1965; Loftsson et al. 2005; Loftsson and Hreinsdóttir 2006). The complex formation is a reversible process:
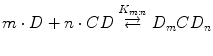
where m drug molecules (D) associate with n cyclodextrin (CD) molecules to form a complex of m:n stoichiometry. K m:n is the observed stability constant of the complex, also known as the binding constant, formation constant, or association constant. The stability constant can be written as follows:
![$$ {K}_{m:n}=\frac{\left[{D}_mC{D}_n\right]}{{\left[D\right]}^m\cdot {\left[CD\right]}^n} $$](http://plasticsurgerykey.com/wp-content/uploads/2017/07/A309277_1_En_14_Chapter_Equ2.gif)
where the brackets denote the molar concentrations. Most commonly, one drug molecule forms a complex with one cyclodextrin molecule:
![$$ {K}_{1:1}=\frac{\left[D/CD\right]}{\left[D\right]\cdot \left[CD\right]} $$](http://plasticsurgerykey.com/wp-content/uploads/2017/07/A309277_1_En_14_Chapter_Equ3.gif)
where, in saturated drug solutions, [D] is the intrinsic solubility of the drug (S 0), i.e., the solubility when no cyclodextrin is present in the aqueous complexation media. The total drug solubility ([D]T) in a given media is then:
![$$ {\left[D\right]}_T={S}_0+\left[D/CD\right] $$](http://plasticsurgerykey.com/wp-content/uploads/2017/07/A309277_1_En_14_Chapter_Equ4.gif)
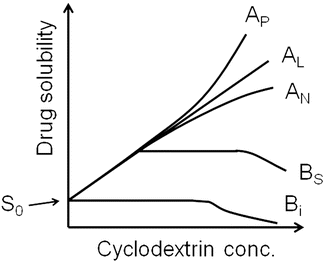
assuming 1:1 D/CD complex formation according to Eq. 14.3. A plot of [D]T versus [CD]T for the formation of a 1:1 D/CD complex should give a straight line (i.e., AL-type phase-solubility diagram, Fig. 14.2) with the y-intercept representing S 0 and K 1:1 defined as (Higuchi and Connors 1965):
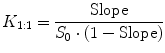
where Slope is the slope of the linear AL diagram. The slope is always less than unity when 1:1 complex is being formed. Complexes of other stoichiometry are less common (Brewster and Loftsson 2007; Loftsson and Brewster 2010). AP-type profile can indicate formation of a complex that is second or higher order with respect to cyclodextrin or that cyclodextrin complex aggregates (nanoparticles) are being formed. The complexation efficiency (CE) is calculated from the slope of the phase-solubility diagram. It is independent of the intercept (or S 0) and frequently used when the influence of various pharmaceutical excipients on the solubilization is investigated (Loftsson and Brewster 2010, 2012). For 1:1 D/CD complexes, the CE is calculated as follows:
![$$ CE=\frac{\left[D/CD\right]}{\left[CD\right]}={S}_0\cdot {K}_{1:1}=\frac{\mathrm{Slope}}{\left(1-\mathrm{Slope}\right)} $$](http://plasticsurgerykey.com/wp-content/uploads/2017/07/A309277_1_En_14_Chapter_Equ6.gif)
The drug:CD molar ratio in a particular complexation media saturated with the drug can thus be calculated from the CE:
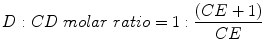
For a more detailed mathematical description of the complex formation, the reader is referred to recent reviews (Brewster and Loftsson 2007; Loftsson and Brewster 2010) and the original publication by Higuchi and Connors (1965). Additionally, the effects of various pharmaceutical excipients on K 1:1 and CE and how they can enhance the solubilizing effects of cyclodextrins have been reviewed (Loftsson and Brewster 2012).
Fig. 14.2
Phase-solubility diagrams. A-type diagrams are due to formation of water-soluble complexes and are usually associated with the water-soluble cyclodextrin derivatives. B-type diagrams indicate formation of poorly soluble complexes that are usually associated with the poorly soluble parent cyclodextrins. S0 is the intrinsic drug solubility, i.e., the solubility of the drug in the complexation media when no cyclodextrin is present
(14.1)
(14.2)
(14.3)
(14.4)
(14.5)
(14.6)
(14.7)
14.2 Cyclodextrins as Permeability Enhancers
In general, chemical penetration enhancers, such as sulfoxides, fatty acids, fatty acid esters, alcohols, amides, and surfactants, enhance drug permeation into and through the skin by permeating into the skin barrier where they temporarily decrease its barrier properties. These penetration enhancers enhance membrane permeation of both hydrophilic and lipophilic drugs and, in most cases, from both nonaqueous and aqueous vehicles. Studies have shown that the permeation-enhancing properties of cyclodextrins are quite different from these chemical permeation enhancers (Masson et al. 1999; Loftsson and Masson 2001; Loftsson et al. 2004; Dahan et al. 2010; Dahan and Miller 2012; Hymas et al. 2012). For example, only negligible amounts of cyclodextrins are able to permeate intact skin and, thus, they do not directly affect the skin barrier. In one study only 0.02 % of topically applied HPβCD was absorbed into intact hairless mouse skin over 24 h period, whereas 24 % was absorbed into stripped skin where stratum corneum had been removed (Tanaka et al. 1995). Another study showed that only 0.3 % of the more lipophilic dimethyl-β-cyclodextrin was absorbed into intact rat skin after topical application (Gerlóczy et al. 1988). In addition, cyclodextrins are only able to enhance drug permeation from aqueous vehicles and in most cases they are only able to enhance permeation of lipophilic poorly water-soluble drugs (Loftsson et al. 2007b, 2008; Loftsson and Brewster 2011; Loftsson 2012).
There are numerous reports on the effects of cyclodextrins on dermal and transdermal drug delivery (Table 14.2). Depending on the experimental conditions and vehicle composition, cyclodextrins either increase or decrease drug permeation through the skin. Still more studies can be found on the effects of cyclodextrins on drug absorption from the gastrointestinal tract and the buccal cavity through the nasal mucosa as well as through other mucosal membranes, all of which can give us some insight into how cyclodextrins act as penetration enhancers (Loftsson et al. 2007b, 2008; Loftsson and Brewster 2011; Loftsson 2012).
Table 14.2
Examples of cyclodextrin-containing dermal formulations and transdermal drug delivery studies
Drug | Cyclodextrin | Reference |
|---|---|---|
Acitretin | RMβCD | Loftsson et al. (1995) |
Alkannin | HPβCD | Chen et al. (1996) |
Avobenzone | HPβCD | Yang et al. (2008) |
Beclomethasone dipropionate | γCD | Uekama et al. (1985) |
4-Biphenylylacetic acid | βCD, DMβCD, HPβCD | |
Bupranolol | HPβCD, PMβCD | Babu and Pandit (2004) |
Capsaicin | HPβCD | Zi et al. (2008) |
Celecoxib | DMβCD | Ventura et al. (2006) |
Curcumin | HPβCD, HPγCD | Hegge et al. (2008) |
Dexamethasone acetate | βCD, HPβCD | Lopez et al. (2000) |
17β-Estradiol | HPβCD | Loftsson et al. (1991) |
Fludrocortisone acetate | γCD | Klang et al. (2012) |
Human growth hormone | αCD, βCD, HPβCD | Shakory et al. (2010) |
Hydrocortisone | βCD, CMβCD, HPβCD, MLβCD, RMβCD | |
Ibuprofen | HPβCD | Iervolino et al. (2000) |
Indomethacin | βCD, DEβCD, DMβCD | |
Ketoprofen | HPβCD | Batzdorf and Mullergoymann (1993) |
Liarozole | HPβCD | Vollmer et al. (1993) |
Lidocaine | DMβCD, HPβCD, SBEβCD | Dollo et al. (1998) |
Loteprednol etabonate | DMβCD | Loftsson and Bodor (1994) |
Melatonin | HPβCD | Lee et al. (1998) |
Metopimazine | MβCD | Bounoure et al. (2007) |
Methyl paraben | HPβCD | Tanaka et al. (1995) |
Miconazole | αCD, HPβCD | Tenjarla et al. (1998) |
Naproxen | βCD | Celebi et al. (1993) |
Piribedil | RMβCD | Legendre et al. (1995) |
Piroxicam | HPβCD | |
Prednisolone | βCD, γCD | Uekama et al. (1987) |
Progesterone | αCD, βCD, γCD | Klang et al. (2010) |
Prostaglandin E1 | αCD, βCD, CMEβCD, | |
Shikonin | HPβCD | Chen et al. (1996) |
Sulfanilic acid | βCD, DMβCD | Okamoto et al. (1986) |
Testosterone | HPβCD | Loftsson et al. (1991) |
Tolnaftate | βCD, βCD-polymer | Szeman et al. (1987) |
Tretinoin | βCD, HPβCD, DMβCD | |
Triamcinolone | HPβCD | Kear et al. (2008) |
14.2.1 Theoretical Background
Drugs permeate the skin via passive diffusion. The driving force for passive diffusion through an aqueous vehicle into the skin and then through the skin is the gradient of chemical potential (μ) (Higuchi 1960; Idson 1971). Likewise, the partitioning of drug molecules from the skin exterior into the outermost skin layer is controlled by the chemical potential. High chemical potential of the drug in topical vehicle is a prerequisite for its good dermal bioavailability:
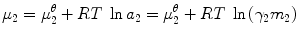
and
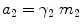
where μ 2 is the chemical drug potential in the vehicle, μ 2 θ is the chemical potential in a given standard state, a 2 is the thermodynamic drug activity, R is the gas constant, T is the temperature in Kelvin, γ 2 is the activity coefficient, and m 2 is the molality of the drug. The thermodynamic definition of the partition coefficient (K o/w) of a drug between organic (o) and aqueous (w) phases is:
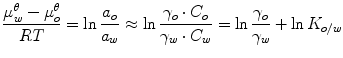
Equation 14.10 states that equilibrium between the two phases is attained when the chemical potential of the drug in one phase (e.g., in water or the aqueous membrane exterior (μ w)) is equal to the chemical potential in the other phase (e.g., the oil phase or the membrane itself (μ o)). Thermodynamic activity is equal to unity in saturated solutions, and, thus, many ointments and creams consist of finely divided drug suspensions. Under such conditions, the vehicle is saturated with drug, and dissolved drug molecules are at their highest potential to leave the vehicle and partition into the skin. Addition of solubilizers, such as cyclodextrins, to an aqueous drug solution will lower the drug activity (i.e., lowers γ w in Eq. 14.10), and, thus, under normal conditions, cyclodextrins lower the potential of the drug to exit the formulation (Másson et al. 2005). However, addition of cyclodextrin to aqueous drug suspension, increasing the amount of dissolved drug while keeping the solution saturated with drug, will not lower the drug activity as long as solid drug is present in the aqueous suspension. Under such condition, the thermodynamic activity (a w in Eq. 14.10) will remain equal to unity, and, thus, dissolved drug molecules are at their highest “exiting” potential, while total amount of dissolved drug is increased. Adding too much cyclodextrin to an aqueous dermal formulation will, on the other hand, decrease the activity (a w) below unity and, consequently, result in less than optimum topical bioavailability. Although passive diffusion is driven by the gradient of chemical potential, it is common to replace it by the concentration gradient. For example, according to Fick’s first law, the driving force for steady-state drug diffusion between two points (i.e., from point 1 to point 2) in a solution is the concentration gradient:
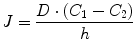
where J is the drug flux, D is the drug diffusion constant, C 1 and C 2 are the drug concentrations at point 1 and point 2, respectively, and h is the distance between the two points.
(14.8)
(14.9)
(14.10)
(14.11)
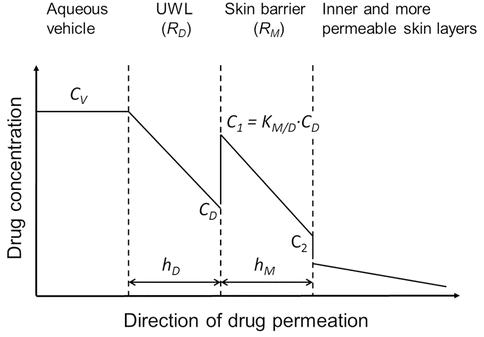
Most biological membranes are multilayer membrane barriers, and most contain various diffusion pathways and transport systems. Higuchi described passive drug transport through multilayer barriers as series of additive resistances analogous to electric circuits (Higuchi 1960). Later drug permeation through biological membranes was described mathematically as drug permeation through a lipophilic membrane sandwiched between unstirred water layers (UWLs) emphasizing that the UWL must be treated as a part of the total membrane barrier (Zwolinski et al. 1949; Flynn et al. 1972; Flynn and Yalkowsky 1972; Loftsson et al. 2007b). Here a simple two-barrier model will be used to explain how cyclodextrins affect drug permeation from an aqueous vehicle into and through the skin or other biological membranes (Fig. 14.3) (Loftsson and Brewster 2011). In this model, the drug molecules encounter two barriers on their way from the vehicle through a lipophilic membrane. The first one is the aqueous boundary layer at the membrane surface, the UWL. The second one is the lipophilic membrane itself, frequently identified as the outermost layer of the skin, stratum corneum. The total skin barrier towards drug permeation consists of the UWL and the lipophilic membrane. Assuming independent and additive resistances of the two layers, the total drug permeation resistance (R T) of this simple membrane can be defined as:
 The Correlation Between Transepidermal Water Loss and Percutaneous Absorption
The Correlation Between Transepidermal Water Loss and Percutaneous Absorption
 Epidermal Lipids and the Intercellular Pathway
Epidermal Lipids and the Intercellular Pathway
 Skin Deep: The Basics of Human Skin Structure and Drug Penetration
Skin Deep: The Basics of Human Skin Structure and Drug Penetration
 The Increasing Importance of the Hair Follicle Route in Dermal and Transdermal Drug Delivery
The Increasing Importance of the Hair Follicle Route in Dermal and Transdermal Drug Delivery
 Liposomal Gels in Enhancing Skin Delivery of Drugs
Liposomal Gels in Enhancing Skin Delivery of Drugs
 Formulation Effects in Percutaneous Absorption
Formulation Effects in Percutaneous Absorption




Related posts:
The Correlation Between Transepidermal Water Loss and Percutaneous Absorption
Epidermal Lipids and the Intercellular Pathway
Skin Deep: The Basics of Human Skin Structure and Drug Penetration
The Increasing Importance of the Hair Follicle Route in Dermal and Transdermal Drug Delivery
Liposomal Gels in Enhancing Skin Delivery of Drugs
Formulation Effects in Percutaneous Absorption
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
