Fig. 9.1
Stratum corneum structure and main penetration pathways through skin (Adapted from Roberts et al. 2008)
Penetrants may cross the skin using a number of routes, i.e. intercellular and transcellular stratum corneum pathways, and appendageal routes (eccrine sweat glands or hair follicles). Whilst it was originally proposed that the transcellular route was the main penetration pathway in skin (Scheuplein and Blank 1971), it is now generally thought that most drugs penetrate via the intercellular pathway (Elias 1983; Barry 2001; Hadgraft 2001). Recent work by Nitsche et al. (Nitsche and Kasting 2008) has suggested that the relative transport through the two pathways is dependent on the nature of the compound. Compounds have long been shown to be taken up into the follicles but this uptake and penetration is believed to be dominant only at early times (Scheuplein and Blank 1971). This hypothesis has been recently confirmed in human skin in vivo by Liu et al., who showed that hair follicles contributed significantly to percutaneous absorption of caffeine only at times soon after application (Liu et al. 2011). Siddiqui et al. (1989) and Essa et al. (2002) have used pharmacokinetic modelling and a skin sandwich technique, respectively, to show that the extent of follicular transport at steady state is between about 5 and 30 % of the total flux. Grice et al. (2010) measured the stratum corneum and the follicles separately and showed that follicular transport for minoxidil was more important at early times. There are a number of conditions such as folliculitis, acne, where there is a need for direct follicular targeting. Topical follicular targeting can avoid systemic toxicity and possibly reduce the dose and frequency of application. The value of the follicular pathway for systemic drug delivery is not well established. Several advantages of follicular drug targeting are (1) bypassing the tortuous pathway of transepidermal absorption, (2) decreasing systemic toxicity and (3) possibly reducing the applied dose and/or frequency of its administration. Recently, there is a considerable interest in using the follicular pathway as a reservoir for localised therapy as well as a transport pathway for systemic drug delivery (Lademann et al. 2008).
9.3 Goals of Skin Delivery
Drugs are applied to skin for local, regional and systemic delivery. The intention of local delivery is to directly treat cutaneous disorders on the skin surface or within the skin directly beneath the application site, such as eczema or psoriasis. Any systemic absorption that may occur is undesirable (Roberts et al. 2002a). In contrast to local delivery, regional delivery requires deposition of the drug with deeper penetration beneath the site of application. Examples of target sites for regional delivery are muscles and joints, beneath and around the application site. Systemic absorption from this form of delivery is undesirable. High concentrations of drug can be localised directly at the target site, thereby reducing systemic drug concentrations and possible systemic side effects (Honeywell-Nguyen and Bouwstra 2005). Systemic delivery requires application of the drug to the skin to treat systemic disorders. Here, the drug should diffuse sufficiently across the skin to achieve therapeutic systemic concentrations (Roberts et al. 2002a). Ideally, there should not be any accumulation of the drug within the skin. As an alternative to oral drug delivery, transdermal delivery avoids gastrointestinal absorption–related problems and hepatic first-pass metabolism and can provide zero-order drug delivery and therefore avoid variability of plasma levels (Honeywell-Nguyen and Bouwstra 2005).
9.4 Mathematical Considerations in Percutaneous Absorption
9.4.1 Principles of Percutaneous Absorption
When a chemical is in contact with skin, quantification of the extent of penetrant absorption through the skin is usually related to Fick’s first law. As has been mentioned before, the stratum corneum is considered as the main barrier to penetration of most drugs and barrier effects in the viable epidermis and dermis are considered negligible. In such cases, Fick’s law relates the amount of solute penetrating across unit area of skin (Q) to the duration of application (t), solute diffusivity (D), effective path length for the diffusion (h), as well as gradient concentration difference between immediately below the area of application C sc(o) and the inner side of stratum corneum C sc(i):
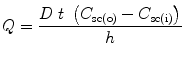
Steady state flux, J ss, is calculated from Eq. 9.1 as
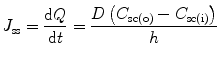
(9.1)
(9.2)
Steady state in Eq. 9.2 is only achieved when the lag time of diffusion has been reached and it is assumed that (i) the skin barrier, usually the stratum corneum, behaves like a pseudo-homogenous membrane with no appreciable changes in its barrier properties; (ii) depletion of the product applied does not occur and (iii) the system has perfect sink conditions (i.e., C sc(i) << C sc(o) and C sc(i) can therefore be disregarded). Accordingly, if the solute stratum corneum-vehicle partition coefficient (K) is the ratio of C sc(o) and the solute concentration in the vehicle C v, and the permeability coefficient (k p) is a product of KD/h, Eq. 9.2 can be written as
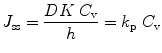
(9.3)
The stratum corneum k p is related to the permeability of the available pathway in the stratum corneum, e.g. intercellular lipid, polar route and transcellular route. For most solutes, it seems that penetration is via the intercellular lipid; therefore, k p usually refers to stratum corneum lipid permeability (Roberts and Walters 1998).
Normally, maximum flux, J max, is achieved at the solute solubility in the stratum corneum (C sc sat) which also corresponds when solute solubility in the vehicle achieves its saturation condition (C v sat). Substituting C sc sat into Eq. 9.2 under sink condition or C v sat into Eq. 9.3 indicates that J max is expressed by either k p and C v sat or by D, h and C sc sat:
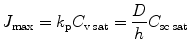
(9.4)
Flux and k p values can be determined practically through in vitro or in vivo studies. In addition, a qualitative structure–penetration relationship approach uses physicochemical properties of the solute in terms of octanol-water partition coefficient (log P) and molecular weight (MW), to give an approximation of k p. Based on published permeability coefficient data from aqueous vehicles, Potts and Guy (1992) generated a model to predict k p (cm h-1) :
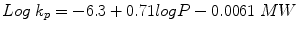
(9.5)
Recognising that the solubility of solutes in water can also be expressed in terms of solute melting point (MP) and Log P, Eq. 9.5 can also be expressed in terms of maximum flux J max (Milewski and Stinchcomb 2012; Pastore et al. 2014, in press):
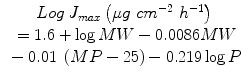
(9.6)
In addition to the stratum corneum being a barrier in skin penetration, other skin layers might also affect the skin penetration process. Equation 9.5 is frequently modified to adjust for the viable epidermal resistance for lipophilic solutes (Cleek and Bunge 1993). Transport in the dermis occurs by both diffusion and convective transport by the blood and the lymphatics. It has been suggested that convective transport will dominate when the compound is highly protein bound and this explains why many non-steroidal drugs provide effective local analgesia after topical application (Dancik et al. 2012; Anissimov et al. 2013). When the viable epidermis or subsequent layers act as extra barriers, Eq. 9.3 for steady state flux can be modified as follows:
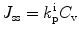
where
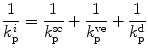
where 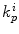 is total skin permeability coefficient and k psc, k pve, k pd represent stratum corneum, viable epidermis and dermis permeability coefficients, respectively. Now, taking into account the overall route available for penetration, then Eq. 9.8 can be modified to account for the permeability coefficient of appendages (k papp) (Jepps et al. 2013):
is total skin permeability coefficient and k psc, k pve, k pd represent stratum corneum, viable epidermis and dermis permeability coefficients, respectively. Now, taking into account the overall route available for penetration, then Eq. 9.8 can be modified to account for the permeability coefficient of appendages (k papp) (Jepps et al. 2013):
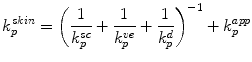
(9.7)
(9.8)
is total skin permeability coefficient and k psc, k pve, k pd represent stratum corneum, viable epidermis and dermis permeability coefficients, respectively. Now, taking into account the overall route available for penetration, then Eq. 9.8 can be modified to account for the permeability coefficient of appendages (k papp) (Jepps et al. 2013):(9.9)
9.4.2 Requirements for Optimised Delivery and Targeting: An Alternative Approach
We have now described the various transport processes required to achieve either topical or systemic delivery. Whatever the intended target of the delivery, solute penetration through the stratum corneum is the prerequisite. During the formulation development process, particularly, the ability to predict the rate of solute penetration through the stratum corneum would be useful to minimise trial and error and shorten the optimisation process. Additionally, it is also highly relevant for assessing potential dermal exposure from industrial and environmental hazards.
Predictive models for percutaneous penetration based on the relationship of steady state flux, solute concentration in the vehicle and skin permeability (k p) are up to now perhaps the most widely studied. They assume that steady state flux is directly proportional to the skin permeability coefficient and the concentration of the penetrant in the vehicle. When the penetrant is at maximum solubility in the vehicle, maximum flux is achieved, provided the system behaves ideally, i.e. no solute-vehicle-skin interactions exist. Under these conditions, flux at a concentration below saturation in a given vehicle can be approximated if the fractional solubility is known.
Conditions required for k p-based validity, however, are not always met. A solute may interact with vehicle to some degree, resulting in an increased or decreased effective solute concentration in the vehicle. When this situation occurs, flux may no longer be directly related to vehicle solute concentration. Another scenario potentially affecting k p-based validity is when either the solute or the vehicle interacts with the stratum corneum, causing alteration in barrier properties. The vehicle may enter the stratum corneum altering solute solubility in the skin. Alternatively, the vehicle may influence stratum corneum lipid packing which in turn may alter solute diffusivity in the stratum corneum.
Another limitation for k p-based prediction may be faced during formulation development or optimisation processes. Given the rapid development in the variety of the potential formulation types in topical and transdermal delivery, a formulator may be eager to compare directly which type of formulation best suits the properties of a given penetrant. The use of k p-based prediction models may be difficult in real formulations where there are a number of solvents, as k p is usually derived from aqueous solutions.
We have recently proposed an alternative way to overcome the limitations of a k p-based approach in predicting the extent of penetration for various different formulations (Wiechers et al. 2012). It is derived from a more fundamental definition where the steady state flux through the SC is directly related to diffusivity (D), path length of diffusion (h) and solute concentration in the stratum corneum (C sc). The solute concentration in the stratum corneum can be measured when the amount of solute retained in stratum corneum and the stratum corneum volume are known. A tape stripping technique can be used to measure the amount of solute retained in the stratum corneum. However, as it may be difficult to estimate the solute concentration in the strips, we have proposed the use of Eq. 9.10, which is a variation of Eq. 9.2:
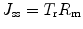
where T r is the rate constant of solute transfer in the stratum corneum, directly related to D/h, and R m is the amount of solute that is retained in the stratum corneum, analogous to C sc. As Eq. 9.10 measures what actually happens in the skin rather than what happens in the formulation, this equation enables us to compare the abilities of different formulations to enhance the skin delivery of solutes. When solutes are saturated in formulations and formulations do not affect the skin, their J ss will be similar. A reduced flux will likely be caused by a lower solute solubility in the formulation (i.e. lower thermodynamic activity in the vehicle) and a higher value will be caused by formulation-skin interactions, to increase either T r or R m. Increased T r can be achieved by increasing diffusion in the membrane, whereas R m can be increased by promoting solute partitioning from the formulation into the stratum corneum.
(9.10)
Given that stratum corneum penetration is the first requirement to exert a local, regional or a systemic effect, an increase in J ss is desirable. However, the relative merit of enhancing either T r or R m depends on the aim of delivery. Topical delivery is best achieved by a localised solute concentration at the target skin layer, with minimal systemic delivery to prevent unwanted systemic side effects. This is achieved by maximising solute retention in the target layer (R mtarget) and the rate of solute transfer to the target layer (T rtarget). For example, when the stratum corneum is the target layer, such as in sunscreens, insect repellents and cosmetic preparations, it would be desirable to have formulations that maximise solute retention in the stratum corneum  , preferably without an increase in T rsc. Similar requirements can be then adopted for the other skin layers. For example, dermal targeting with anaesthetic formulations requires high solute transfer that can facilitate solute diffusion to the dermal layer (i. e. high T rsc, T rve and T rdermis) and high solute retention in the epidermal/dermal layers to localise solute accumulation R mdermis. Systemic delivery is best achieved by formulations that can facilitate solute diffusion across the skin to achieve systemic therapeutic solute concentrations. In other words, this requires high T rskin with optimized R mskin (R mskin = R msc + R mve + R mdermis).
, preferably without an increase in T rsc. Similar requirements can be then adopted for the other skin layers. For example, dermal targeting with anaesthetic formulations requires high solute transfer that can facilitate solute diffusion to the dermal layer (i. e. high T rsc, T rve and T rdermis) and high solute retention in the epidermal/dermal layers to localise solute accumulation R mdermis. Systemic delivery is best achieved by formulations that can facilitate solute diffusion across the skin to achieve systemic therapeutic solute concentrations. In other words, this requires high T rskin with optimized R mskin (R mskin = R msc + R mve + R mdermis).
, preferably without an increase in T rsc. Similar requirements can be then adopted for the other skin layers. For example, dermal targeting with anaesthetic formulations requires high solute transfer that can facilitate solute diffusion to the dermal layer (i. e. high T rsc, T rve and T rdermis) and high solute retention in the epidermal/dermal layers to localise solute accumulation R mdermis. Systemic delivery is best achieved by formulations that can facilitate solute diffusion across the skin to achieve systemic therapeutic solute concentrations. In other words, this requires high T rskin with optimized R mskin (R mskin = R msc + R mve + R mdermis).9.4.3 Skin Pharmacokinetics: Drug Concentration in the Target
In relation to the goal of skin delivery, the concentration at the target site could be mathematically modelled based on the related transport process. In the viable epidermis, other processes that may affect solute penetration and distribution within the skin include epidermal metabolism, solute binding and sequestration. These variables may also affect solute availability (F) for solute penetration into the dermis and beyond. The steady state solute concentration at a site C ss is approximated by accounting for input rate and local clearance (Anissimov et al. 2013; Jepps et al. 2013). In general, the concentration at any skin layer target site is determined by the steady state flux input to the target site 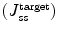 , the bioavailability (F), the area of application (A) and the local clearance (Cl local):
, the bioavailability (F), the area of application (A) and the local clearance (Cl local):
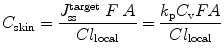
, the bioavailability (F), the area of application (A) and the local clearance (Cl local):(9.11)
For systemic delivery, the concentration of solute in plasma after topical application, C ssp, can be approximated from Eq. 9.11, in which k pskin is defined as in Eq. 9.9:
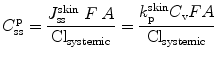
(9.12)
Equation 9.12 can be used in finding the best drug candidate for achieving the required J sstarget based on drug physicochemical properties, as shown in Table 9.1.
Table 9.1
Effective plasma concentration, clearances, required steady state flux and physicochemical data used to predict required solute transdermal flux for passive topical delivery systems (Pastore et al. 2014, in press)
Solute | Plasma level (ng/mL) | Cl systemic (L/h/70 kg) | Estimated J ss required (μg/h) | t 1/2 (h) | MW | MP (°C) | Log P |
|---|---|---|---|---|---|---|---|
Clonidine | 0.2–2.0 | 15 | 3–30 | 8–13 | 230 | 130 | 2.7 |
Estradiol | 0.04–0.06 | 600–800 | 24–48 | ~1 | 272 | 173–179 | 4.2 |
Fentanyl | 1–3 | 27–75 | 27–225 | 3–12 | 337 | 83–84 | 3.9 |
Nicotine | 5–30 | 77 | 385–2,310 | 2 | 162 | −79 | 1.1 |
Nitroglycerin | 0.02–0.4 | 216–3,270 | 4.32–1,308 | 0.03–0.05 | 227 | 13 | 1 |
Scopolamine | >0.05 | 65–121 | 3.25–6.05 | 1–5 | 303 | 55 | 0.8 |
Testosterone | 3–10.5 | 41 | 123–430.5 | 0.17–1.7 | 288 | 155 | 3.6 |
9.5 Drug Candidates
Despite the advantages of using skin as a drug application site, to the present only a few drugs are marketed commercially in transdermal delivery systems. Some applications include motion sickness, hypertension, angina, anaesthesia, hormone replacement therapy, contraception and smoking cessation therapy (Finnin and Morgan 1999; Thong et al. 2007). This limited application is due mainly to the limited permeability of the stratum corneum. Improved understanding of percutaneous transport and skin structure has led us to conclude that the best candidate drugs for passive skin delivery must have certain physicochemical properties: low molecular weight (<500 Da), moderate lipophilicity, melting point <250 °C and good potency (daily systemic dose ≤20 mg) (Finnin and Morgan 1999).
Studies on Quantitative Structure–Penetration Relationships (QSPR) (Anderson and Raykar 1989; Magnusson et al. 2004) have shown that solute size is important for facilitating the diffusion process while adequate lipophilicity is required to provide sufficient solubility in the stratum corneum. Maximum flux was shown to have an inverse relationship with molecular weight (Magnusson et al. 2004) and in aqueous vehicles reached a maximum when 2.7 < log P <3.1 (Zhang et al. 2009). However, it does not necessarily follow that all lipophilic solutes with log P > 3 are poor candidates for skin delivery, because drug potency must also be considered. Lippold (Wenkers and Lippold 2000) tested the correlation between maximum flux and potency of a range of anti-inflammatory drugs with varying lipophilicities and showed diclofenac, with a log P > 4, to have the highest efficacy coefficient ratio (the ratio of maximum flux and drug potency dose). Even though the maximum flux of diclofenac was lower than drugs with log P in the range 2.7–3.1, its potency was greater, since the anti-inflammatory effect of a drug also depends on lipophilicity. A similar concept was also recognised for anti-mycotics as shown in Fig. 9.2 (Mertin and Lippold 1997). It is observed that nystatin is an inappropriate compound because it has a low J max due to its large size and at the same time is not very potent. Griseofulvin also shows poor efficacy due to both poor penetration and a higher MIC. The most ideal compound for efficacy appears to be amorolfine, which has a lower J max than naftifine but is much more potent (lower minimum inhibitory concentration or MIC).
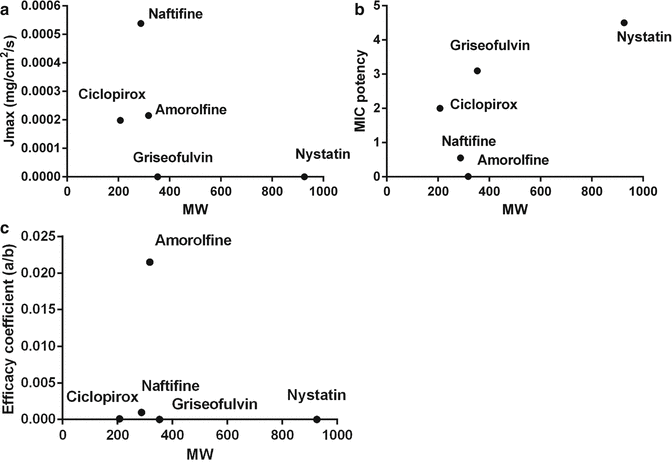
Fig. 9.2
Efficacy concept for antimycotics, whereby efficacy (c) is defined as the ratio of its maximum penetration flux (J max, a) from a formulation to its potency defined in terms of its minimum inhibitory concentration (MIC, b) (Mertin and Lippold 1997)
9.6 Formulation-Based Strategies for Optimised Percutaneous Absorption
Formulation encompasses multiple processes that lead eventually to a successful product in the market. The demonstration of product efficacy and good customer acceptance would perhaps be key points for achieving such a goal. While in a broader sense, dosage form design, composition design, packaging design and industrial scaling-up are also essential parts of the formulation process, we will limit our discussion in this chapter to the choice of potential techniques and ingredients that can be adopted to optimise dermal and transdermal drug delivery.
Based on the theoretical requirements described in previous sections, improved skin delivery of a solute can be achieved by increasing its concentration in the stratum corneum and its rate of transport in this barrier. Table 9.2 summarises formulation-based passive current approaches for percutaneous absorption enhancement and their possible effect on R m and/or T r. Other aspects considered later include thermodynamic activity, chemical enhancers, hydration, ion pairs, complex coacervation, pH adjustment and eutectic mixtures.
Table 9.2
Formulation-based enhancement strategies and their possible effects on solute diffusivity and retention in stratum corneum
Approaches | Mechanisms | Potential effects |
|---|---|---|
Thermodynamic activity | Supersaturation | Increase partitioning; may increase R m |
Formulating for Efficacy | Increase partitioning and/or R m; (potentially) increase D | |
Chemical penetration enhancer | Stratum corneum lipid alteration, e.g. fluidised lipid bilayer, lipid extraction, polar head group alteration, hydrophobic lipid | Increase D |
Stratum corneum desmosomes and protein alterations | Increase D | |
Corneocytes alteration | Increase D | |
Shift stratum corneum solubility closer to that of permeant | Increase R m and/or partitioning | |
Increase solute solubility in the stratum corneum because of solvent-drag mechanism | Increase partitioning and/or R m | |
Ion pairs and complex coacervates | Increase lipophilicity | Increase partitioning |
Eutectic mixture | Decrease melting point | Increase R m |
9.6.1 Escaping Tendency from Formulation (Thermodynamic Activity)
The thermodynamic activity of a solute can be estimated from the ability of the solute to escape from the formulation (i.e. its vapour pressure). Thermodynamic activity also has been referred to as the effective solute concentration in the applied formulation. Increasing the solute concentration is a common way to increase thermodynamic activity. When saturation conditions are reached, the condition in which the solute has maximum thermodynamic activity, the skin delivery rate will be the highest. This fundamental concept was clearly shown by Twist and Zatz (1986) with their flux comparison of saturated paraben from various solvents through a silicone membrane. The fluxes were similar, despite the concentration difference of paraben due to the variation of solubilities. As thermodynamic activity is based on the extent of solute solubility in the applied vehicle/formulation, the degree of solubility or saturation provides a useful way of comparing solute thermodynamic activity among different vehicles/formulations. Percutaneous absorption is directly related to the thermodynamic activity unless the skin is affected by solute– or vehicle–skin interactions (Twist and Zatz 1986).
The thermodynamic activity concept has been used to develop formulations for many years (Poulsen et al. 1968; Ostrenga et al. 1971; Woodford and Barry 1982; Ishii et al. 2010). For example, Poulsen et al. (1968) optimised the composition of propylene glycol-water to modulate the release of 0.025 % fluocinolone and its ester. It was shown that release of both of the drugs was maximal at the least amount of propylene glycol needed to completely dissolve the drug, i.e. 20 and 75 % of propylene glycol in water for fluocinolone and fluocinolone ester, respectively. A high proportion of propylene glycol in water beyond the required least amount to completely dissolve steroids increased the solubility of both steroids and thus decreased the partition coefficient, leading to lower thermodynamic activity. A dependency of drug release on the vehicle and the nature of the drug could be demonstrated. Similar results have been observed by Ostrenga et al. (1971), who found that a relationship could be built between in vitro and in vivo data and emphasised the importance of thermodynamic activity considerations in the development of formulations.
Another example of the role of thermodynamic activity as a determinant of skin penetration flux was reported by Barry et al. (1985a, b). In their work, the thermodynamic activity of benzyl alcohol, a volatile hydrogen-bonding model penetrant, was measured using headspace chromatography from various binary solvents. The vapour flux across human abdominal skin was directly related to its thermodynamic activity (Barry et al. 1985b). However, when liquid flux was tested, the relationship was less consistent. It was found that some liquid vehicles, such as toluene, damaged the skin (Barry et al. 1985a). This result indicates that if skin is affected by vehicle-skin interactions, thermodynamic activity and flux relationship may not be linear. Expanding on the thermodynamic activity approach, concepts such as Formulating for Efficacy and Supersaturation have been developed and applied.
9.6.1.1 Formulating for Efficacy (FFE)
This concept was first introduced by the late Johann Wiechers in 2004 (2004). FFE was developed to combine two contradictory properties required of by a formulation, namely, (i) the formulation has maximum solubility for the active chemical to accommodate the required dose and (ii) the formulation should have, at the same time, minimum active chemical solubility so as to promote the active chemical leaving the formulation and partitioning into the stratum corneum. A Relative Polarity Index (RPI) is used to measure the polarity difference between the active ingredient and the vehicle. To achieve the first goal, a solvent or emollient is chosen based on a low RPI. Because the first chosen solvent or emollient has similar polarity to that of the active chemical, they are mutually soluble; however, the escaping tendency is low. Therefore, a second solvent or emollient is added to the formulation to reduce the active chemical solubility to just above the required solubility to solubilise the therapeutic dose. This second solvent or emollient has a large RPI. Wiechers recently published a practical application of the principles of FFE based on maximum flux considerations (Wiechers et al. 2012).
9.6.1.2 Supersaturation
Supersaturation is a condition in which an active chemical has greater thermodynamic activity than that occurring under saturation conditions. A supersaturated formulation can be designed by different methods, including co-solvent mixing. The major drawback for this system is its instability. Crystallisation may occur during storage, leading to a change in therapeutic performance of the formulation. An anti-nucleating agent such as a polymer is often added to reduce crystallisation. Supersaturated conditions can also sometimes be formed after application of the formulation to the skin surface. Volatile components such as ethanol or water may evaporate, leading to supersaturated conditions in the remaining formulation (Pellett et al. 1994, 1997). As seen in in vitro (Oliveira et al. 2012) and in vivo experiments (Stinchcomb et al. 1999), there was an initial rapid solute uptake when volatile solvent evaporated from the formulation, leading to increasing solute concentration and eventual supersaturation. However, after complete evaporation, penetration was markedly decreased as the solute was deposited as a crystallised film on the skin surface.
9.6.2 Chemical Penetration Enhancers
Chemical penetration enhancers are purposely incorporated in topical or transdermal formulations to alter solute skin solubility, partitioning from the formulation to the skin and/or diffusivity in the stratum corneum for promoting skin delivery. Ideally, they should also be non-irritant, cosmetically acceptable (e.g. odourless, tasteless and colourless), rapid acting with predictable duration, reversible and chemically stable. Potential mechanisms by which solvents and vehicles, commonly used as enhancers, may affect stratum corneum properties have been summarised in earlier work (Roberts et al. 2002b). Briefly, altered skin barrier properties may result from the extraction of stratum corneum lipids, the penetration of enhancers into or through the stratum corneum and their interactions with stratum corneum components (Lane 2013). Either intercellular lipids or intracellular proteins of the stratum corneum, as proposed in the Lipid-Protein-Partitioning theory of Barry (1991) could be potential sites of action for enhancers. The effects of these interactions may alter stratum corneum-vehicle partitioning, skin solubility and/or penetrant diffusion in the stratum corneum.
There is no accepted consensus on the classification of enhancers. Nevertheless, simple classifications such as organic vs. inorganic or polar vs. non-polar have been used. In this review, enhancers are grouped according to the potential mechanisms by which enhancers affect D, K, R m or combinations of those parameters.
9.6.2.1 Effects on Solute Diffusivity
Enhancers have been postulated to affect diffusivity in the stratum corneum in various ways. They can act on stratum corneum lipids as well as proteins. They may disturb the lipid organisation of the stratum corneum, making it fluidised. As the result of the disturbed stratum corneum lipid packing, D may be increased. Enhancers may act on the hydrophobic stratum corneum lipid tails and create free volume available for a penetrating solute to diffuse. Examples of common enhancers that work to increase diffusivity are laurocapram (Azone®, Whitby Research Inc., USA) and oleic acid. They may be dispersed in the stratum corneum intercellular lipid, such as Azone® (Hoogstraate et al. 1991), or may be located in separate domain pools within lipid domain, such as oleic acid (Ongpipattanakul et al. 1991). Terpenes such as limonene and cineole have also been shown to increase drug diffusivity (Moghimi et al. 1996a, b). In addition to disturbance of the stratum corneum lipid packing, solute diffusivity may also be increased as a result of stratum corneum lipid extraction. Some solvents such as dimethyl sulphoxide (DMSO) and ethanol may act by this mechanism although more systematic research needs to be conducted to confirm the mechanism (Barry 2001; Benson 2005). Alternatively, enhancers such as polar solvents may also interact near the polar head groups of the stratum corneum lipids. Interactions with keratin in the corneocytes may open the dense protein structure and thereby increase D. Surfactants, DMSO and urea interact with keratin. The peptide/protein in the lipid bilayer domain may also be affected by this type of enhancer (Benson 2005).
9.6.2.2 Effects on Solute Partitioning and Solubility in Stratum Corneum
Enhancers that increase solute solubility in the stratum corneum can promote partitioning into the skin and hence improve flux. If enhancers permeate into the stratum corneum and shift the skin polarity closer to that of the permeant, the permeant skin solubility and hence partitioning will increase (Cross et al. 2001). A similar result may be expected via a solvent-drag mechanism, in which vehicle entering into the stratum corneum carries solute with it (Zhang et al. 2009; 2011). A number of solvents have been reported to increase partitioning and stratum corneum solubility, e.g. ethanol, propylene glycol, Transcutol® and N-methyl pyrrolidone (Williams and Barry 2004; Benson 2005).
9.6.2.3 Synergistic Effects on Diffusivity and Solute Skin Solubility
Combinations of enhancers which increase D and K or stratum corneum solubility have been reported to give a synergistic effect of enhancement. Examples for this approach are Azone®/propylene glycol for metronidazole (Wotton et al. 1985) and Azone®/diethylene glycol monoethyl ether (Transcutol®, Gattefosse Co., France) for prostaglandin (Watkinson et al. 1991) as well as oleic acid and propylene glycol (Walker and Smith 1996; Benson 2005).
9.6.2.4 Formulation Dependency of Enhancement Effects
It is important to note that enhancers may affect skin properties through different mechanisms, resulting in either enhancement (Megrab et al. 1995; Watkinson et al. 2009), retardation (Hadgraft et al. 1996) or sometimes no significant effect (Sheth et al. 1986). The effect may be either drug dependent (Aungst et al. 1986), concentration dependent (Megrab et al. 1995; Watkinson et al. 2009), time dependent (Grice et al. 2010) or vehicle or formulation dependent (Sheth et al. 1986; Rhee et al. 2007). Sheth et al. (1986) compared the penetration enhancement of trifluorothymidine (TFT), an anti-viral compound, amongst propylene glycol, polyethylene glycol-300 (PEG 300) and water with and without the addition of Azone®. Without Azone®, TFT flux was ranked in the order of propylene glycol > water > PEG 300. With the addition of 5 % Azone®, TFT flux increased three- to fourfold as the ratio of PG : PEG 300 in the vehicle went from 0:100 to 100:0. This suggested that the extent of enhancement was dependent on the vehicles in which the enhancers were incorporated. In addition, it seemed that PEG 300 was not a good solvent for enhancement purposes (Hadgraft 1983).
Related posts:
 The Correlation Between Transepidermal Water Loss and Percutaneous Absorption
The Correlation Between Transepidermal Water Loss and Percutaneous Absorption
 Epidermal Lipids and the Intercellular Pathway
Epidermal Lipids and the Intercellular Pathway
 Skin Deep: The Basics of Human Skin Structure and Drug Penetration
Skin Deep: The Basics of Human Skin Structure and Drug Penetration
 The Increasing Importance of the Hair Follicle Route in Dermal and Transdermal Drug Delivery
The Increasing Importance of the Hair Follicle Route in Dermal and Transdermal Drug Delivery
 Liposomal Gels in Enhancing Skin Delivery of Drugs
Liposomal Gels in Enhancing Skin Delivery of Drugs
 Formulation of Drug-Cyclodextrin Complexes
Formulation of Drug-Cyclodextrin Complexes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


