2006 versus 2007 (%)
2005 versus 2006 (%)
2004 versus 2005 (%)
Collagen
−60
−27
−72
Hyaluronic acids
−9
+33
+35
During 2009, law firm McPhadden Samac Tuovi announced a proposed class action against Johnson & Johnson and related companies relative to the injectable collagen dermal filler Evolence. It is alleged that Johnson & Johnson and related companies failed to warn the public about the potential of these adverse effects. Exactly, during November 2009, Johnson & Johnson announced that it would be discontinuing the porcine collagen product.
The need of a double test and relative waiting time together with the risk of side effects are probably the main reasons leading to a gradual decrease in the mid of 2000s and a definitive loss of popularity of heterologous collagen between 2008 and 2010.
1.2.2 Human Collagen
Implantable materials containing autogenous or isogenous human collagen have a limited presence because preparation of the injectable solutions is relatively difficult, and the cost of these products is relatively high.
1.2.3 Autogenic Human Collagen
Introduced at the end of the 1980s, Autologen (Collagenesis, Inc., Beverly, MA) was the first autologous injectable agent on the market. Autologen is a dispersion of intact collagen fibres and a matrix of collagen tissue obtained from the clean skin of the patient during a plastic surgery procedure (mammaplasty, abdominoplasty, facelift and blepharoplasty). A skin biopsy is inadequate. Because the injected material is autologous and no allergic reactions were reported in a sufficient number of patients, the United States Food and Drug Administration (FDA) does not consider it necessary to perform a test before Autologen injection. The skin excision, placed in a sterile container, is sent to the manufacturer’s laboratory for treatment. As a general rule, 10–13 cm2 of excised skin is required to produce 1 mL of Autologen 5 %.
At least 3 injections, a few weeks apart, are needed to obtain a satisfactory result, provided that each treatment is overcorrected by 30 % [17]. The duration of treatment depends on the region treated, the injection technique and the volume of Autologen administered. No significant side effects have been reported. It must be noted, however, that moderately severe erythema may last for 48 h after the injection [18, 19]. Preparation of the autologous collagen from a patient-tissue sample is expensive, and yield varies, depending on the individual and the anatomic areas from which collagen is harvested. Since 1998, autologous fibroblast cultures have been used to correct wrinkles, scars and other skin defects. Boss et al. [20] described a method of injecting autologous fibroblasts obtained from a 3-mm skin excision from the retroauricular area, an area protected from UV light. The sample is immediately placed in a culture medium provided by Isolagen Laboratories (Houston, TX) and must reach the laboratory by the day after sampling in an isothermic container. Six weeks after sampling, an injection test (0.1 mL) is administered to the patient in the forearm; any sign of an allergic reaction is recorded. Two weeks after the test, approximately 1 mL of the autologous material is available for implantation. Overcorrection of 300 % is recommended for suitable aesthetic results [21, 22]. The level of correction achieved depends on the defect, the patient’s age, and the ability of the patient’s fibroblasts to create collagen. Patients older than 60 years are not good candidates for this technique. This technique has several disadvantages. The Isolagen preparation must be injected within 48 h. It offers more effective correction of periorbital wrinkles or perioral wrinkles than of deep furrows [23]. The improvement obtained is poor compared with that of other techniques, such as hyaluronic acid, and correction is not immediate. Fagien and Elson [24] concluded that the results obtained with this technique were rather disappointing. In addition, Isolagen is expensive.
1.2.4 Isogenic Human Collagen
Alloderm (LifeCell, Branchburg, NJ) has been used in the treatment of burns and for transplantation in periodontal surgery. In aesthetic surgery, it is used to increase lip volume, to correct nasolabial folds and to treat scars. Alloderm is an acellular dermal graft material obtained from cadavers or from a tissue bank that provides an acellular matrix of dermal components, including collagen, elastin and glycosaminoglycans. The dermis skin is examined in accordance with FDA requirements and regulations relating to human tissue. No cases of transmissible viral disease have been reported in patients who have received this treatment since its introduction in 1992. Alloderm is offered in the form of sheets that are implanted through an incision in the treatment area. Infection of the incised/sutured sites has been attributed to abscess formation around the suture rather than to the graft itself. Cases of labial herpes have also been reported; prophylactic antiviral therapy must be prescribed for patients with a history of labial herpes. Alloderm is also available in a micronized injectable form, marketed under the name Cymetra (average particle size 123 μm). Cymetra is provided in the form of an aseptic powder reconstituted with lidocaine 0.5 % with 1: 200,000 epinephrine immediately before injection. It is injected with a 26-gauge needle [25]. Dermalogen (Collagenesis Corp., Beverly, MA) is obtained from cadaver tissues that have been carefully selected to help eliminate the risks of viral and bacterial infection. Clinical indications for the use of Dermalogen include correction of obvious nasolabial folds, perioral wrinkles, glabellar wrinkles and depressed scars, as well as increasing lip volume. The injection technique for Dermalogen is the same one used for Autologen: injection into the middle dermis/deep dermis with a 30-gauge needle. Because the injection is painful, use of a local anaesthetic is recommended [26]. Overcorrection of 20–30 % is recommended at each session. An average of 3 injection sessions is required for satisfactory correction. Prolonged erythema and acneiform rashes were noted in 10 % of patients in a study of 130 patients. The manufacturer does not recommend a pretreatment allergy test, although 1 case of foreign-body reaction 4 weeks after a test injection of 1 mL of Dermalogen in the forearm has been described by Moody and Sengelmann [27]. Klein [28] also reported several positive skin tests with Dermalogen and 1 case of secondary reaction characterized by redness, swelling, and hyperpigmentation of the treated sites after Dermalogen implantation.
1.3 Hyaluronic Acid
Chiara Lumini
Hyaluronic acid is a natural substance in the body that was first described in 1934 by John Palmer and Karl Meyer at Columbia University, New York. It was isolated from a cow’s eye, and the name comes from hyalos (Greek word for glass) and the uronic sugar found in the substance. Hyaluronic acid is produced by the cells in the human body, and it plays a key role in numerous functions. It facilitates the cell-division process, it makes the skin elastic and it lubricates the joints. In 2003, the U.S. Food and Drug Administration (FDA) approved the first hyaluronic acid dermal filler for the correction of moderate to severe facial wrinkles and folds, such as nasolabial folds. In the last ten years, numerous products for therapeutic and aesthetic uses have been developed for this versatile natural substance.
1.3.1 What is Hyaluronic Acid?
Hyaluronic acid is a natural complex sugar found in all living animals. It is one of the few elements that is virtually identical in all living organisms. Hyaluronic acid has the capacity to bind great quantities of water, absorbing more than 1,000 times its weight In addition, hyaluronic acid combines with collagen and elastin. The triple binding of elastin, collagen and hyaluronic acid provides elasticity and volume to the skin.
The body’s amount of hyaluronic acid is metabolized quickly and must be newly produced constantly by the cells.
The ageing of the skin, and the exposure to oxidants, to pollutants and to ultraviolet rays, reduce the ability of the cells to produce hyaluronic acid. As a result, the skin begins to reduce its volume, with subsequent formation of facial wrinkles and folds.
1.3.2 What is Cross-Linked Hyaluronic Acid?
In its natural form, hyaluronic acid is a liquid composed of individual polymers (chains) that are broken down in the body in just 12 h. Cross-linking is a process in which the individual chains of hyaluronic acid are cross-linked (chemically bound) together, converting the liquid hyaluronic acid into a gel, a soft solid. The hardness of the gel depends on the degree of cross-linking of the individual hyaluronic acid chains.
The body metabolizes cross-linked hyaluronic acid more slowly than the natural individual chains, resulting in a longer duration of effect.
For this reason, dermal fillers composed of cross-linked hyaluronic acid are used in aesthetic medicine to temporarily replace lost hyaluronic acid and to restore volume of the face and of the body.
Fillers are reticulated hyaluronic acid-based medical devices. They are made of stabilized non-animal hyaluronic acid obtained from bacterial fermentation of_Streptococcus equi strains.
The cross-linking agent generally used for fillers is 1.4-butanediol diglycidyl ether (BDDE), a small molecule that binds to two ends of an HA chain, generating a three-dimensional structure.
The various HA-based fillers on the market have different chemico-physical characteristics, such as particle size, the cross-linking agent used, the degree of cross-linking, the quantity of free HA and the elastic modulus G.
The quantity of hyaluronic acid in a product affects its consistency and longevity. However, it is also important to take into account the quantity of reticulated HA, the quantity of non-reticulated HA and the degree of cross-linking to consider the HA as fully or partially linked. Often, some quantities of free hyaluronic acid are added to increase its ease of injection as it functions as a lubricant.
The cohesive property of the gel depends on the elastic modulus G which measures its resistance to deformation and, therefore, the strength of the gel. Fillers with a high G are considered cohesive gels and are indicated for correction of deeper wrinkles, such as naso-labial folds or for marionette lines, whereas fillers with a lower G′ are indicated for the treatment of larger areas such as the cheekbones and cheeks (See Table 1.2).
Different properties of hyaluronic acid can be resumed in a simple classification:
low viscosity hyaluronic acid, useful for revitalization and small wrinkles
medium viscosity hyaluronic acid: useful for deeper wrinkles, lines and lips
high viscosity hyaluronic acid: useful for larger areas as malar areas.
1.3.3 History of Hyaluronic Acid in Aesthetic Medicine
1987 Q-Med was founded by Bengt Ågerup with a view to commercializing the research that he had carried out around hyaluronic acid.
1996 Restylane® obtained marketing authorization in Europe for use as filler for wrinkles and lip augmentation.
2003 Restylane® is approved in the US.
2003 Corneal group introduces Juvaderm in Europe and Canada.
In 2006, Q-Med launches a new product for lip enhancement treatment, Restylane Lipp™, Q-Med’s first product in the field of body augmentations, Macrolane™, is approved in Europe.
The JUVÉDERMTM dermal filler family of products is approved by the FDA.
2007 Restylane Perlane™ approved for sale in the USA.
2008 Q-Med’s Restylane Vital™ and Restylane Vital™ Light—were launched in Europe.
Allergan introduces in Europe Juvederm Ultra 2, 3 and 4.
2009 Restylane obtained registration approval in China and became the first injectable non-animal hyaluronic acid product on the Chinese market.
Q-Med introduced Restylane® Lidocaine and Restylane Perlane™ Lidocaine.
Allergan introduces Juvaderm Voluma.
2010 By the beginning of 2010 FDA approved Restylane and Restylane Perlane with added lidocaine in USA.
2011 Belotero Balance has been approved by FDA to treat moderate to severe facial wrinkles and folds. De-listing of Q-Med AB (publ) from NASDAQ OMX Stockholm. Q-med is acquired by Galderma.
CE-approval of Restylane SubQ lidocaine.
2012 Glytone Range of Injectable Hyaluronic Acid Products of Pierre Fabre Group was acquired by Merz Aesthetics Inc.
Allergan introduces the new filler Juvederm Volbella with lidocaine.
1.4 Macrofillers: Hyaluronic Acid for Body Modelling
Giulia Beltrami
The use of liquid filler for body enhancement was first described in 1899 by Gersuny, who injected silicone into the scrotum of a patient in order to reconstruct a testicle after an orchiectomy [29, 30]. Since then, there has been a steady increase in the use of implants and filler substances to reshape body defects. In particular, many permanent liquids and gels have been injected for breast augmentation (e.g. silicone, paraffin, polyalkylimide and polyacrylamide hydrogel). However, the use of these materials has been hampered by complications, such as chronic inflammatory reactions, palpable nodule formation, granuloma formation, and migration [29, 31–33]. As a consequence of these complications, new materials on the market can face scepticism, even if they are biocompatible and non-permanent.
Hyaluronic acid specifically produced for body remodelling was introduced in 2009, in particular Macrolane, a new formulation of injectable stabilized hyaluronic acid–based gel of non-animal origin (NASHA-based gel; Q-Med AB, Uppsala, Sweden) have been developed for use in breast enhancement, volume restoration and contouring body surfaces [31, 34]. Since larger volumes are required for body enhancement than for facial augmentation, the Macrolane formulation has increased viscosity (i.e. a thicker gel). With a high resistance to deformation, hyaluronic acid gel augments body tissue simply by occupying space (in a similar manner to permanent implants).
In 2010, Bioscence (Bioscence, Germany) introduced Hyacorp for body enhancement, with exclusion of breasts. Hyacorp is a specially designed cross-linked HA gel which becomes less viscous under pressure (injecting force) and returns immediately to the original viscosity upon pressure release (injecting force). This is a characteristic of a system (advanced thixotropic technology = ATT technique) exhibiting a decrease in viscosity with an increase in the rate of shear, usually a function of time.
Breast augmentation was Macrolane major indication. Many women have undergone breast augmentation with Macrolane in Europe and Asia. It has never been approved for breast augmentation by the Food and Drug Administration [31, 34]. In January 2010 Goisis et al. wrote a contribution to Aesthetic Plastic Surgery [35]. Three major questions were examined in this paragraph. The first question is related to the duration and cost of Macrolane. Macrolane is a resorbable material, and the study of P. Heden et al. published in 2009 showed 30–50 % resorption at 12 months [31]. The echographic measurements in the study of Goisis et al. [35] showed a 60 % rate of resorption after one year. Consequently, a second treatment was usually done 9 months after the first. The necessity of a touch-up increases the cost of breast augmentation with NASHA gel, which is as expensive as a single surgical treatment with a prosthesis after three or four touch-ups. The second problem is related to the radiological evaluation of patients. Macrolane is a new material and it constitutes a diagnostic challenge for radiologists. Indeed, the appearance of NASHA gel may mimic a cyst on mammography and sonography [31]. Therefore, it is important that radiologists become familiar with the spectrum of imaging findings of Macrolane. In particular, in order to make an accurate diagnosis in these patients, it is important to send them to specialized radiological centres. The last open question is related to post-treatment adverse events. Goisis et al. had a high rate of minor complications. Goisis et al. concluded in this study that NASHA gel for breast volume augmentation was an interesting treatment, although the three open questions require more studies. P. Heden et al. answered this paper with a letter, writing that Macrolane injection did not reduce sensitivity and specificity in breast cancer screenings [36]. After the paper of Goisis et al, some other authors reported complications associated with the procedure [32, 37]. In 2011, Goisis et al. wrote two papers about difficulties on breast imaging, which were described by manyauthors as the most important problem of breast augmentation by hyaluronic acid [38–40]. To reduce these problems, Goisis et al. stressed the importance to carry out the injections under ultrasound guidance. In particular, they suggested to inject a single deposit of hyaluronic acid gel. A cavity with a uniform symmetrical globular shape must be created in the space under the deep fascia of the breast, anterior to pectoralis muscle. The gel can be also placed posterior to the pectoralis muscle (submuscular plane) or inside the pectoralis muscle (intramuscular plane) in order to minimize visibility and palpability. In fact, in Goisis et al. published experience, the overall incidence of complications among patients receiving submuscular or dual-plane or intramuscular injection appeared lower than in patients receiving subglandular injection. Injection of gel into the space between the glandular tissue and the deep fascia of the breast may be related to a higher risk of displacement into the gland tissue and into the subcutaneous fat, especially if multi-pocket injection is performed without ultrasonography guidance [41]. In April 2012, after reviewing the current situation and in consultation with the regulatory authorities, Q-Med has decided to discontinue the indication for breast augmentation until consensus for best practices in breast radiology examination following Macrolane treatment has been reached. Q-Med announced that a safety reporting system has been in place since launch and no safety concerns have been identified. Despite this, Macrolane can interfere with the reading of mammograms. Since no safety concerns have been identified with the product itself, Q-Med suggested that women who have undergone breast augmentation with Macrolane do not need to take any additional actions other than the routine follow-up consultations or as directed by their doctors. As with any other breast prosthesis, it is important to inform the healthcare professional of previous Macrolane treatment before any breast assessment is carried out.
1.5 Calcium Hydroxylapatite
Gina Bianco
Initially launched in the US in 2000 as Radiance FN, this dermal filler was introduced in Europe in June 2004 under the name Radiesse™. Radiesse™ is a medical device developed by Bristol-Myers Squibb and BioForm Medical (since acquired by Merz Aesthetics).
Radiesse is an injectable filler material composed of synthetic calcium hydroxylapatite microspheres (30 %) suspended in an aqueous carrier gel (70 %). These uniform microspheres (25–45 m) are smooth in shape and are identical in composition to the mineral portion of human bone and teeth [42].
Radiesse injectable implant is a steam sterilized, latex-free, non-pyrogenic, semi-solid, cohesive completely biodegradable deep and sub-dermal implant. The principle component is synthetic calcium hydroxylapatite. Calcium hydroxylapatite is the primary mineral constituent of bone and teeth. The semi-solid nature of the implant is created by suspending calcium hydroxylapatite in a gel carrier that consists primarily of water and glycerine. The gel structure is formed by the addition of a small amount of sodium carboxymethyl cellulose. It does not contain any animal- or human-derived components. The gel is dissipated in vivo and replaced with soft-tissue growth, while the calcium hydroxylapatite remains at the site of injection.
Results from extensive in vitro and in vivo safety studies and in several retrospective physician reports, including toxicology assessments, standardized biocompatibility testing, and a 3-year animal study, demonstrate that injectable calcium hydroxylapatite is biocompatible, non-toxic, non-irritating and non-antigenic. Patient sensitivity testing is not required before use [43].
Calcium hydroxylapatite has been used for more than 20 years in various forms in surgery and dentistry. In the United States, injectable calcium hydroxylapatite has been used for several years for the correction of oral/maxillofacial defects, for vocal fold augmentation and as a radiographic tissue marker. In 2006, Radiesse was approved in the United States for the correction of moderate to severe facial wrinkles and folds, including the nasolabial folds, and restoration and/or correction of the signs of facial fat loss (lipoatrophy) in people with human immunodeficiency virus. In Europe, Radiesse™ is approved for plastic and reconstructive surgery, including deep-dermal and sub-dermal soft-tissue augmentation of the facial area.
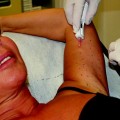
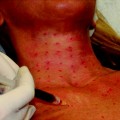
When placed into soft-tissue, Radiesse provides immediate correction. Over time, the carrier gel is gradually absorbed and the calcium hydroxylapatite particles remain (Figs. 1.1, 1.2, 1.3). Local histiocytic and fibroblastic response at the site appears to result in the production of new collagen around the microspheres [44]. Preclinical canine studies (Fig. 1.4, above) have demonstrated histologically progressive integration of collagen fibres in and around the calcium hydroxylapatite microspheres up to 78 weeks after implantation (Fig. 1.4, centre and below).
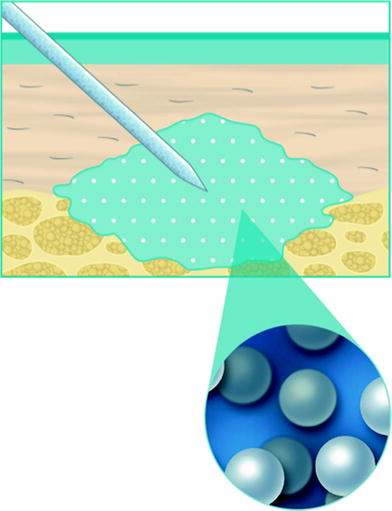
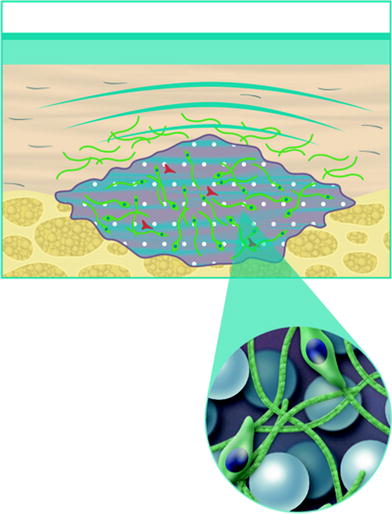
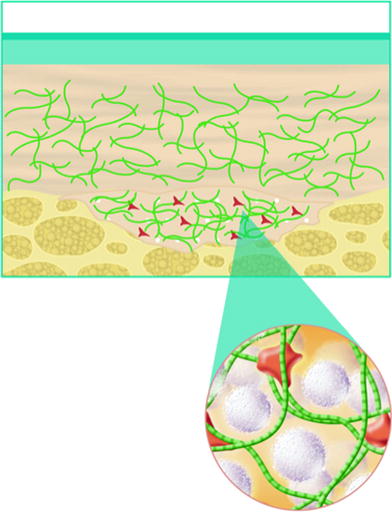
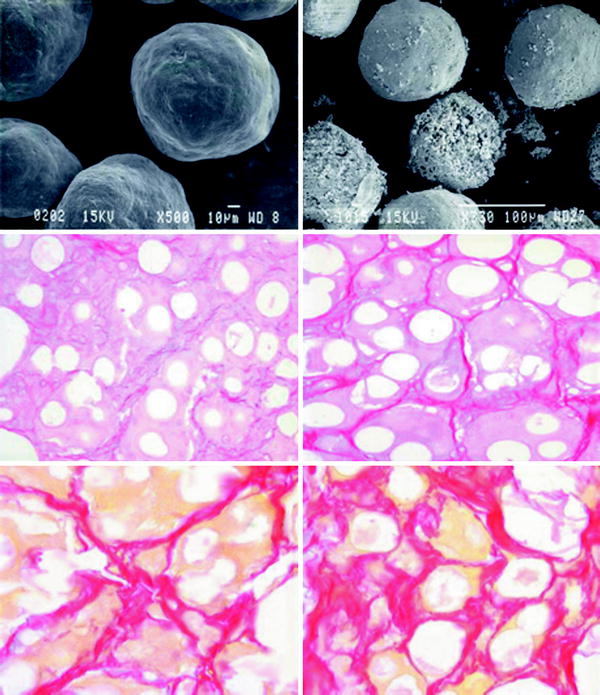
Fig. 1.1
When injected into soft-tissue, Calcium hydroxylapatite provides immediate correction
Fig. 1.2
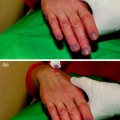
Over time, the carrier gel is gradually absorbed and the calcium hydroxylapatite particles remain
Fig. 1.3
Local histiocytic and fibroblastic response at the site appears to result in the production of new collagen around the microspheres
Fig. 1.4
Scanning electron photomicrographs of (above, left) calcium hydroxylapatite microspheres in sodium carboxymethylcellulose gel. Note the consistent, smooth, round shape (500 magnification). Above, right Calcium hydroxylapatite microspheres 30 months after implantation into bladder neck (350 magnification). Note the slowly dissolving particles. Centre and below Histological evaluation of calcium hydroxylapatite gel injected intradermally into canine skin over a period of 4–78 weeks. Sections stained with picrosirius red denote progressive collagen integration in and around the white spherule calcium hydroxylapatite particles. Collagen deposition (centre, right) 16 weeks, (below, left) 32 weeks, and (below, right) 78 weeks after calcium hydroxylapatite injection (Photographs and illustrations courtesy of BioForm Medical, Inc.)
Over time, calcium hydroxylapatite particles are broken down into calcium and phosphate ions via normal metabolic processes and eliminated through the body’s normal excretory processes. In one long-term animal study in the bladder neck, the particles remained intact at the site of injection throughout the entire 3-year study period [45]. Our experience with calcium hydroxylapatite use for facial soft-tissue augmentation has shown results lasting an average period of a year or more in most patients. In vivo, durability depends on factors such as injection technique, site of material placement and patient age and metabolism. The study was conducted on 357 patients and is not yet published.
1.5.1 Injection Technique
Due to its relatively low viscosity, calcium hydroxylapatite is delivered with a small (e.g., 27-gauge) needle and local anaesthesia improves patients’ comfort during procedures.
The choice of infiltration, nerve block anaesthesia, topical anaesthesia, infiltration of tiny amounts of local anaesthetic directly into the area, or some combination thereof depends on the preferences of the operator and the patient [46, 47]. When needed, the treatment site should be marked before administration of anaesthetic, as infiltration may distort the skin surface.
Calcium hydroxylapatite should ordinarily be injected at the sub-dermal plane, especially when filling creases, wrinkles and deep lines. Injection depth can be just in the subcutaneous space but superior to the periosteum. The injection can also be placed on the periosteum if the intent is to augment the facial bony skeleton. Placement on the periosteum will not stimulate bone growth in the area. Depending on the area being treated, calcium hydroxylapatite may be injected in a retrograde fashion using a linear, threading, fanning and/or crosshatching technique. Massage or moulding of material follows injection to desired effect. Post-treatment care involves immediate placement of ice onto the injected areas to reduce and limit tissue oedema and ecchymosis.
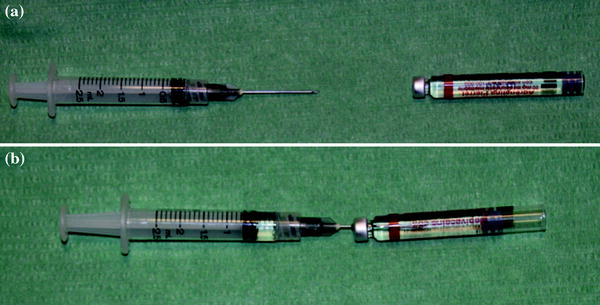
Practical skill: a lesser volume of calcium hydroxylapatite is required to provide the same degree of correction as hyaluronic acid and collagen
Fig. 1.5
a, b 0.5 cc of local anesthetic (2% lidocaine or 2% mepivacaine) are inspired in a 2.5 cc syringe
Fig. 1.6
a, b 1, 5 cc of Radiesse is mixed with the anaesthetic solution
Two studies support the finding of smaller volumes in calcium hydroxylapatite than in several other soft-tissue fillers. For example, in a split-face study of calcium hydroxylapatite versus collagen for the nasolabial folds, on average, the collagen-treated side of the face required twice the volume of material (2.35 ml) to produce optimal correction as compared with the calcium hydroxylapatite–treated side 1.22 ml) (p 0.0001) [48]. In another study, approximately 30 % less volume of calcium hydroxylapatite was required than hyaluronic acid for full correction of the nasolabial folds [49].
1.6 Botulin
Claudio Rinna, Chiara Brillante
1.6.1 History
Botulinum toxin (BoNT) is a neurotoxin. The symptoms of intoxication by botulinum were first described two centuries ago by Justinus Kerner [50, 51]. He observed these symptoms after eating sausages, and the term botulism derived from the latin botul (sausage).
BoNT is produced by the anaerobic micro-organism Clostridium botulinum. There are 7 different known serological types, indicated with the letters from A to G [51]. In 1937, the ophthalmologist Alan B. Scott used for the first time the botulinum toxin A (BTXA) in experiments on monkeys. In 1946, Edward Schantz in Fort Detrick produced the first botulin toxin in crystals [50].
Only in the 1970s, it was approved as a therapeutic agent [51], and in 1989, the BTX-A was approved by the Food and Drug Administration for the treatment of strabismus, blepharospasm and hemifacial spasm in patients over the age of 12. In 1991, it was used to treat spasticity, both in adults and in children and in 2000 for the treatment of cervical dystonia. Jean Carruthers first describes in 1996 the use of botulin toxin for aesthetic treatment of the face.
In 2002, the use of BTX-A in aesthetic medicine was approved for the temporary improvement of glabellar frown lines [52].
The use of BoNT, which has been approved, varies widely depending on the type of toxin, the type of marketed product and the country, even within the European Union. This means that, in some cases, its clinical use is off-label, even if supported by scientific literature. BTXA is marketed in the form of various pharmaceutical products: Botox, Dysport, Xeomin, Vistabex, Bocoture, etc. BoNT B is instead marketed under the name of Neurobloc or myobloc.
1.6.2 Different Toxins
Botulinum neurotoxins are synthesized together with different haemagglutinins and/or non-toxic non-haemagglutinin proteins that form a protein complex called progenitor toxin (See Table 1.2). The protein complex is used to stabilize and protect neurotoxin from degradation. The size and composition of nontoxic proteins in the complex vary depending on the bacterial strain. Different bacterial strains also produce different serotypes of boluninum toxin, known as types A, B, C1, D, E, F and G. The various serotypes form protein complexes of different sizes, in which only the type A forms the larger complex of approximately 900 kd. In all complexes, the portion of active neurotoxin is approximately the same size (150 kd) [53].
The neurotoxin is first synthesized as a single-chain protein that, in order to be active, must be cleaved or affected by the protease. The proteolytic cleavage yields a double-chain active molecule consisting of a heavy chain (approx. 100 kd) and a light chain (approx. 50 kd).
At present, different medicinal products are registered and marketed and although they are all based on type A BoNT, they are manufactured according to different biological processes and obtained through different isolation and purification processes. Each type of BoNT has its own unit of measurement and the different units of measurement are not interchangeable.
In 2009, the US Food and Drug Administration established that BoNTs should be identified unequivocally by means of a specific common denomination for each medicinal product marketed in the United States [52].
BoNTs have been named as follows:
onabotulinumtoxin A (equivalent to the medicinal product BOTOX or Vistabex);
abobotulinumtoxin A (equivalent to the medicinal product Dysport or Azzalure);
incobotulinumtoxin A (equivalent to the medicinal product Xeomin or Bocouture);
rimabotulinumtoxin B (equivalent to the medicinal product Myobloc or Neurobloc in Europe).
This need arose from the consideration that, since BoNT is a product of biological origin, its biological activity, defined potency, cannot be determined with a standard methodology. In fact, potency should be determined for each product lot and, due to its high sensitivity, must be precisely determined and calculated during the entire production process of the toxin itself, from the bulk to the final lot.
Potency is measured as units of biological activity using the test lethal dose 50 (DL50) defined as the quantity of a substance, per unit of body weight, that will kill 50 % of test animals.
In recent years, Allergan Inc. has announced the approval of a test based on in vitro cells to be used for testing the stability and potency of the Allergan BoNT, thereby eliminating the need to perform tests on animals. This new assay is the first to be developed and approved and specifically applies to the type A BoNT manufactured by Allergan.
A different name allows to confirm that the BoNTs are manufactured according to different biological processes, are obtained with different isolation and purification techniques and derive from different strains of the C. Botulinum.
The different formulation and molecular structure can affect both the local diffusion of the toxin from the inoculation site and its potency characteristics which may affect its efficacy, safety profile as well as the antigenic potential of the product.
The BoNT produced by Allergan derives from the clostridium strain called ‘Hall’ which undergoes several purification and crystallization steps (using the method developed by Schantz) which allow to obtain a homogenous neurotoxin complex with a molecular weight of 900kDA given by the presence of auxiliary proteins (haemagglutinins and non-haemagglutinins) that form complexes with the neurotoxin and are used to stabilize and protect the toxin from degradation [54].
As regards the other available preparations, one derives from the clostridium strain called NCTC 2916 which undergoes purification steps using chromatography, a technique that has been proven to generate a more heterogeneous compound, while the other contains pure neurotoxin with a molecular weight of 150Kda, free from complexing proteins [55, 56].
1.6.3 Use in Clinical Practice and Aesthetic Medicine
BoNTs used in aesthetic medicine are of type A and are indicated for the treatment of expression wrinkles (dynamic wrinkles). The effect of proper treatment with BoNT is to mitigate these signs at rest without affecting the mimicry of the patient or inhibiting muscles contraction.
1.6.4 Clinical Evaluation of the Patient
Before the injection of the toxin it is important to perform a careful physical examination of the upper third of the face considering asymmetries, position of the eyelids and the presence of ptosis produced by the levator muscle of the eyelid. To exclude a ptosis, ask the patient to look forward, to put a hand on his forehead to block the frontal muscle in order to evaluate the actual activity of the levator muscle of the eyelid.
The next step is to assess wrinkles in a sitting position and during mimic efforts look for possible contraindications. Once the suitability of the subject to injection has been established, photograph the face both in static and dynamic position to have a documentation prior to treatment.
To proceed with the injection, it is necessary to inform the patient of the possible risks of side effects and let him/her sign an informed consent to treatment.
1.6.5 Contraindications to Treatment
Contraindications to treatment with BoNT -A are:
diseases of the neuromuscular junction (myasthenia gravis, Lambert–Eaton syndrome);
allergy to human albumin and sodium chloride;
skin lesions of treated areas;
infection of the planned site for the injections;
recent surgical treatments;
pregnancy and lactation;
recent use of aminoglycoside antibiotics or other agents that interfere with neuromuscular transmission (spectinomycin, tubocurarin-type muscle relaxants).
1.6.6 Informed Consent
It is important to inform the patient about the good rules to follow during the hours after treatment, such as avoiding alcohol, avoiding excessive pressure on the affected area and exposure to excessive heat.
The patient should be informed of possible side effects from the injection of BoNT, such as pain, weakness, itching, swelling or bruising at the injection site.
The patient should be informed of the side effects that may occur in the hours following the treatment to enable him to associate the disorder to treatment and inform their doctor if necessary. These effects are divided into categories based on the frequency with which they occur:
Very common (1 in 10 patients): headache, local muscle movement disorders and feeling of heaviness in the upper part of the face;
Common (1–10 in 100 patients): eyelid oedema, eyelid ptosis, eyelid inflammation, eye pain, blurred vision, nausea, dizziness, dry mouth, cramps and muscle contraction, flu-like symptoms, itching and burning sensation in the treated area, infection, sensitivity to light, bronchitis, dry skin, fatigue, insomnia, depression;Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree