, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
25.1 Connective Tissue Nevus
25.1.1 Clinical Features
Connective tissue nevi frequently are present at birth but may be acquired. They occur sporadically or may be a feature of certain genodermatoses such as Proteus syndrome, Buschke Ollendorff syndrome, multiple endocrine neoplasia type 1, and tuberous sclerosis [1]. For example, the cerebriform connective tissue nevus is a pathognomonic feature of Proteus syndrome, and it is characterized by an expansive furrowed plaque, most often on the soles of the feet. Incidence and prevalence of these lesions are not known.
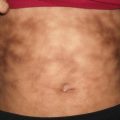
Connective tissue nevi are skin-colored dermal plaques frequently with overlying epidermal thickening, cobblestoned or cerebriform changes described as “pigskin” or “peau d’orange” (Fig. 25.1). They may be solitary or multiple with symmetric distribution at the back, buttocks, and extremities [1]. Connective tissue nevi are persistent. However, excision is usually not required as lesions are often asymptomatic and not cosmetically noticeable.
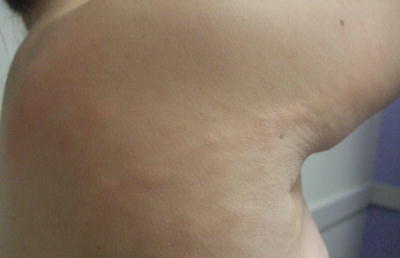
Fig. 25.1
Connective tissue nevus presents as symmetric skin-colored, subcutaneous plaques in linear bands along the upper back
25.1.2 Histology
Various hamartomatous proliferations of connective tissue fall under the term connective tissue nevus and these will be described separately. However, in many cases, the patterns are mixed.
Collagenoma is the most common type of connective tissue nevus, and is characterized by a proliferation of thickened, haphazardly-oriented dense and coarse collagen bundles throughout the dermis [2] (Figs. 25.2 and 25.3). In some cases, electron microscopy has demonstrated increased number of myofibroblasts admixed with the dense collagen bundles [3]. A trichrome stain highlights the haphazard orientation of the dermal collagen bundles, and an elastic tissue stain may demonstrate fewer elastic tissue fibers within the lesional skin. In many cases, the diagnosis is subtle, and it is easiest to establish when lesional skin is compared with a biopsy taken from adjacent, clinically normal skin.
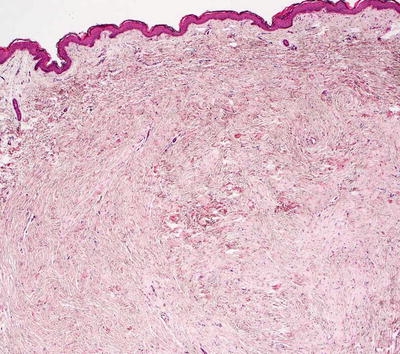
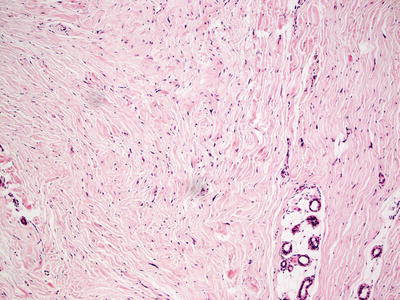
Fig. 25.2
A collagenoma is a type of connective tissue nevus that demonstrates a nodule of thickened collagen bundles haphazardly oriented with some bundles lying perpendicular to the surface of the skin
Fig. 25.3
Increased bundles of thickened and poorly oriented collagen are seen in collagenoma . Decreased elastic tissue fibers as seen on an elastic tissue stain are also a feature (not shown here)
Elastoma is a hamartomatous proliferation of elastic tissue fibers. Increased elastic tissue fibers, which may appear thickened and clumped, may be readily apparent on routine histologic sections. However, the changes may not be visualized without the aid of elastic tissue stains, in which case the elastic tissue changes are easily detected [4] (Fig. 25.4). Structural anomalies within elastic tissue fibers have been observed by electron microscopy [5].
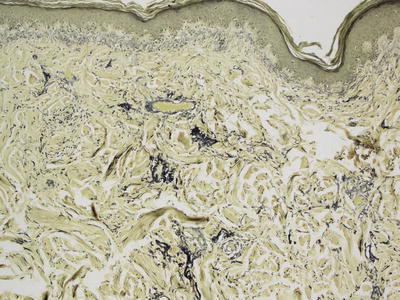
Fig. 25.4
An elastoma is another form of connective tissue nevus characterized by increased number and abnormal clumping of elastic tissue fibers as seen on Verhoeff-van Gieson (VVG) stain
A recently described variant of connective tissue nevus has been named a mucinous nevus. In one case, overlying epidermal acanthosis and hyperkeratosis indicate that the mucin present in the superficial portion of the dermis occurred in concert with an epidermal nevus [6]. In other cases, typical changes of the collagenoma type of connective tissue nevus have been described, in addition to the epidermal nevus and dermal mucin [7].
A rare type of connective tissue nevus called fibroblastic connective tissue nevus demonstrates a poorly circumscribed accumulation of fibroblasts admixed with thickened and haphazardly-oriented collagen bundles. These lesions are located in the deep reticular dermis and may extend into the subcutis. Clusters of bland-appearing fibroblasts are seen in intersecting bundles and may entrap appendages [8]. The fibroblasts are CD34+, similar to those seen in dermatofibrosarcoma protuberans. It is important to be aware of this rare entity in order to prevent over diagnosing a malignant proliferation .
25.1.3 Pathogenesis
Connective tissue nevi are cutaneous hamartomas characterized by an abnormal proliferation of collagen, elastin, or proteoglycans [1, 9, 10]. They can occur sporadically, or in association with genetic syndromes, such as Proteus syndrome, which is characterized by progressive, mosaic, and segmental overgrowth. As mentioned above, the cerebriform connective tissue nevus is a key cutaneous marker of Proteus syndrome [11, 12] .
25.2 Keloid and Hypertrophic Scar
25.2.1 Clinical Features
Keloids are most commonly seen in those with darkly pigmented skin. There is a reported incidence of 4.5 % in Hispanic populations and 16 % in those of African descent [13]. Peak occurrence is between puberty and 30 years of age with rare appearance in infancy [13]. Keloids also may appear or expand during pregnancy.
Hypertrophic scar may be more common than keloids [13]. The propensity for both hypertrophic scar and keloid formation appears to be partially familial. These scars most commonly occur in wounds under tension as well as in body locations with a higher density of melanocytes. Early lesions may be erythematous, while older lesions tend to be more hyperpigmented. Keloids are smooth, firm nodules or plaques with a rubbery consistency that do not respect the borders of the original wound margins. This is in contrast to hypertrophic scars which demonstrate boundary limitation (Fig. 25.5). Patients may experience associated pruritus or pain. While various treatment options exist, keloids have greater incidence of recurrence than hypertrophic scars, which often resolve spontaneously [13].
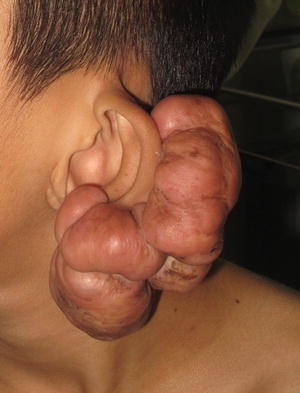
Fig. 25.5
Erythematous, firm nodular plaque at the pinna of the ear extends beyond the original wound margins in a patient with a keloid scar (photo courtesy of Minh Hoang, MD, Ho Chi Minh City, Vietnam).
25.2.2 Histology
The histologic changes in keloid include a dense overgrowth of markedly thickened and eosinophilic collagen bundles in the dermis (Figs. 25.6 and 25.7). The collagen bundles are often densely packed with less than the expected amount of interstitial ground substance. In early lesions, increased fibroblasts may be present, but in well-developed lesions, the dermis appears relatively hypocellular. Virtually no elastic tissue fibers are identified within keloids [14]. The thin vertically-oriented vessels usually seen in well-formed scars may be present, but they can be obscured by thickened collagen bundles.
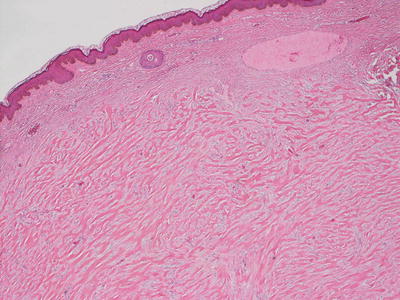
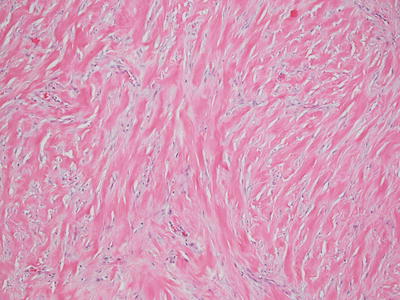
Fig. 25.6
A keloid demonstrates abundant thick eosinophilic bands of collagen that are easily observed at low magnification
Fig. 25.7
Keloidal collagen bundles are haphazardly oriented and can be perpendicular to the skin surface
The histologic findings of a hypertrophic scar include a proliferation of fibroblasts in a nodular configuration (Figs. 25.8 and 25.9). Neovascularization is present and blood vessels are often vertically oriented within the dermis. The epidermis may be flattened with loss of rete ridges, as well as loss of appendages in the areas of fibroblastic proliferation. The cellularity of the lesion varies with its age. In early hypertrophic scar, there are abundant small vessels and dermal fibroblasts, while in late stage lesion the number of cells in the dermis is much lower. The fibroblasts retain an orientation that is parallel to the surface of the skin in most cases. Elastic tissue fibers are markedly diminished within the area of new collagen formation.
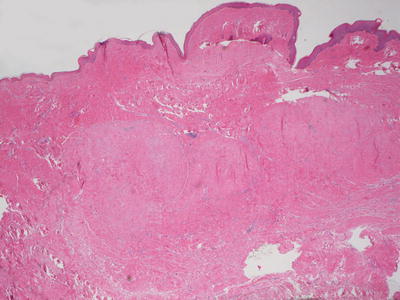
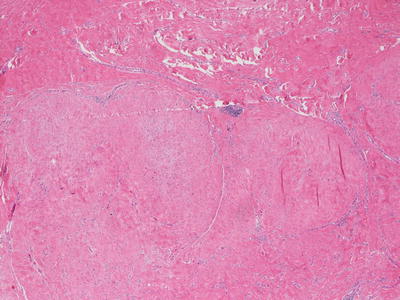
Fig. 25.8
A hypertrophic scar demonstrates a lobular appearance within the dermis. The epidermis is often flattened and there is loss of cutaneous appendages in the region of the scar
Fig. 25.9
Lobules of new collagen can be distinguished from the background native collagen in a hypertrophic scar, and may resemble the lobularity seen in a leiomyoma
The nodular configuration in a hypertrophic scar may raise the differential diagnosis of a leiomyoma, although a trichrome stain or immunostains to identify smooth muscle actin can easily make this distinction. A keloid demonstrates much thicker and more eosinophilic bundles of collagen, but scattered bundles of keloidal collagen may be seen in small quantity within a hypertrophic scar .
25.2.3 Pathogenesis
Keloid scar is a result of dysfunctional wound healing in the skin with excess deposition of extracellular matrix materials. Altered lipid metabolism may play a role in the pathogenesis of keloid scar. Keloids express high levels of arachidonic acid, which is a precursor of pro-inflammatory eicosanoids, such as leukotrienes, prostaglandins, and thromboxanes [16]. It has been proposed that lipids are important in the chronic inflammatory processes that drive keloid development [17].
Systemic hypertension has been suggested to play a role in keloid scar. Hypertension-associated changes in pathological scars are mediated in part by inflammation, hypoxia, and the angiotensin/renin–angiotensin–aldosterone system [18]. Hypoxia can promote fibrosis by stimulating an epithelial-to-mesenchymal transition that is important in fibrosis [19, 20]. Stem cells may play a role in keloid scar formation [21]. Mesenchymal-like stem cells have been identified in keloid tissues [22–24]. These stem cells exhibit the potential to differentiate into cells of bone, cartilage, fat, and endothelial cell lineages.
Interleukin-6 (IL-6) is an inflammatory cytokine important in tissue fibrosis. IL-6 is highly expressed in keloid scar as compared with normal skin [25, 26]. IL-6 induction may contribute to the formation of keloids by promoting the proliferation and differentiation of keloid precursor cells [21].
Hypertrophic scar may be caused by an imbalance between excessive extracellular matrix formation and inadequate matrix remodeling [27]. Hypertrophic scar results from a prolonged inflammatory response to injury, leading to increased vascularization, hypercellularity, excessive collagen deposition, and a decrease in small leucine-rich proteoglycans [28, 29]. Molecular signals, such as stratifin secreted by keratinocytes, exert downstream effects on skin fibroblasts and the production of extracellular matrix proteins [30]. Bone marrow–derived mesenchymal stem cells have been identified in hypertrophic scar and may play a role in scar formation [31].
Hypertension has been linked to pathological fibrosis and scarring, including hypertrophic scar [18]. Hypertension results in increased activation of transforming growth factor (TGF)-β and tumor necrosis factor (TNF)-α signaling [32]. Hypertrophic scar demonstrates an increased number of fibroblasts compared to normal skin and increased activity of TGF-β, which results in overproduction of collagen by dermal fibroblasts [33]. Fibroblasts in scar can differentiate into myofibroblasts, which can also account for increased collagen and extracellular matrix production [34].
Studies have shown that TSG-6 (TNF-α Induced Protein 6), which is an anti-inflammatory cytokine, is important in hypertrophic scar formation. TSG-6 can suppress inflammation and reduce the extent of hypertrophic scar formation in an experimental animal model [35]. TSG-6 reduces the formation of hypertrophic scar by decreasing the synthesis of collagens I and III [35]. TSG-6 inhibits the expression of the inflammatory cytokines IL-6 and IL-1β as well as neutrophil infiltration [35, 36]. Given these anti-inflammatory effects, TSG-6 may be a new therapy for hypertrophic scar treatment .
25.3 Dermatofibroma
25.3.1 Clinical Features
Dermatofibromas are common tumors, accounting for 3 % of the cutaneous specimens assessed by one dermatopathology laboratory in a published report [37]. Dermatofibromas are most frequently seen in young and middle-aged adults and more common in females [37]. Lesions present as a well-defined, hyperpigmented and firm dermal nodule that dimples with lateral compression (“dimple sign”). They may occur anywhere on the body, but are most commonly seen on the lower extremities (Fig. 25.10). Dermatofibromas are typically solitary, although multiple lesions have been reported in 10 % of affected individuals [37]. Dermatofibromas are benign but can persist following onset. They are amenable to surgical excision, and can recur if not completely removed [38].
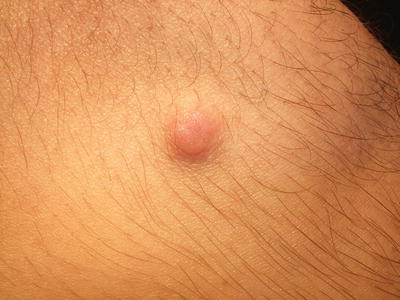
Fig. 25.10
Dermatofibroma presents as a firm erythematous papulonodule that dimples with lateral compression on the thigh of a teenage boy
25.3.2 Histology
The classic dermatofibroma demonstrates a proliferation of fibroblasts underlying an acanthotic and hyperpigmented epidermis (Fig. 25.11). The rete ridges are elongated, and there is increased basilar melanin. In some cases, follicular induction can give rise to a basilar proliferation that resembles basal cell carcinoma. Within the dermis, most cases demonstrate a slight zone of sparing within the papillary dermis. Beginning in the superficial reticular dermis and extending throughout the dermis but not usually into the subcutis, fibroblasts are present in a whorled or storiform pattern around collagen bundles (“collagen trapping”) (Fig. 25.12). The lesion sometimes extends into the fibrous septa between the fat lobules, but ordinarily does not involve the subcutaneous lobules. The cells are somewhat pleomorphic with spindle-shaped cells admixed with plumper cells with abundant cytoplasm. Multinucleated cells may be present. The cells may contain variable amounts of cytoplasmic lipid, and in some cases, extensive lipidization may be apparent [39]. Scattered mitoses can be seen. Collagen bundles separate the individual fibroblasts that do not grow in a fascicular pattern. At the periphery of the lesion, keloid-like collagen may be apparent with interspersed fibroblasts. Older lesions may demonstrate zonation with cellular areas at the periphery and a more sclerotic region present in the center.
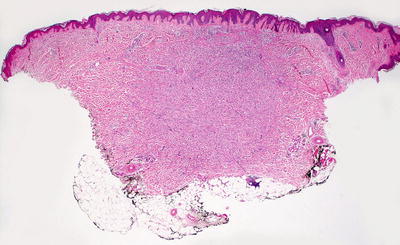
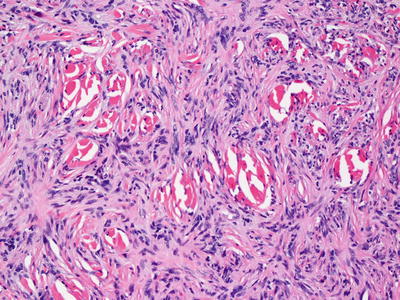
Fig. 25.11
An acanthotic and hyperpigmented epidermis overlies a proliferation of spindle-shaped cells in the dermis in dermatofibroma
Fig. 25.12
A storiform pattern of spindle-shaped cells surrounds and isolates collagen bundles (collagen trapping) in dermatofibroma
Several variants of dermatofibroma share the overall architectural pattern with the classic form but have distinctive histologic characteristics that lead to subtyping with minor clinical relevance [40]. The aneurysmal or angiomatoid variant demonstrates abundant large, ectatic blood vessels admixed with narrow vascular channels. A myxoid variant shows areas with abundant myxoid matrix in the setting of a dermatofibroma. Another variant demonstrates keloidal collagen. A variant with abundant eosinophils has also been described [41].
The major differential diagnoses include dermatofibrosarcoma protuberans (DFSP) , dermatomyofibroma, and xanthogranuloma. The spindle cell proliferation in DFSP often extends up right to the base of the epidermis unlike the zone of sparing that characterizes most dermatofibromas. DFSPs are generally more cellular, and have a more uniform appearance within the spindle cell population that course within interlacing fascicles. The proliferation often extends into the subcutis, and envelops individual adipocytes within the lobules. While scattered mitoses are present, there is minimal cytologic atypia. Minimal collagen is present between tumor fascicles, unlike the case with dermatofibroma. Dermatomyofibroma is a dermal-based proliferation of spindle shaped cells that generally have more abundant eosinophilic cytoplasm than is seen in dermatofibroma. The proliferation tends to grow in a plate-like pattern than is parallel to the surface of the skin. Immunostains for smooth muscle actin, calponin, and muscle-specific actin label the majority of the spindle-shaped cells in dermatomyofibroma [42]. Xanthogranuloma enters the differential diagnosis of a lipidized dermatofibroma. This distinction is generally not one of clinical importance, and in some cases the overlap is so great as to prevent a distinction [43]. The angiomatoid variant may be confused with Kaposi’s sarcoma, but can be distinguished on the basis of areas that resemble a classic dermatofibroma .
25.3.3 Pathogenesis
Dermatofibroma is a benign fibrohistiocytic proliferation. It has some features of a reactive tissue process as well as a proliferative neoplastic process. Some studies have proposed that dermatofibroma is a local response to inflammation or trauma, in which there is an inflammatory response with granulation tissue formation, followed later by a reparative fibrous response [44]. Other studies have indicated that dermatofibroma is a neoplastic process due to its clonal proliferative growth [45]. Multiple dermatofibromas can occur in association with systemic lupus erythematosus [46].
Molecular factors such as syndecan-1, fibroblast growth factor receptor 2, and transforming growth factor-β might be inducers of fibrosis in dermatofibroma [46–49]. Mast cells, which are present in high numbers, may be a cellular source of these fibrinogenic factors. Fibroblasts derived from dermatofibroma lesions are more responsive to IL-1α and IL-1β than fibroblasts derived from normal skin [50]. IL-1 may play a role in fibroblast proliferation in an autocrine manner. Gene fusions involving protein kinase C gene have been identified in some cases of dermatofibroma [51], while rearrangement of ALK gene has been identified in epithelioid and atypical variants [52, 53].
25.4 Dermatomyofibroma
25.4.1 Clinical Features
Dermatomyofibroma is a rare benign cutaneous tumor of mesenychmal derivation. It is most commonly diagnosed in young women [54]. However, in pediatric cases there is no sex predilection. Dermatomyofibroma is described as reddish-brown plaque or nodule, most often occurring on the neck and upper trunk (Fig. 25.13) [54]. It responds well to surgical excision with no reports of metastasis or recurrence. Interestingly, dermatomyofibroma in male children may involute spontaneously, while continued growth is seen in females .
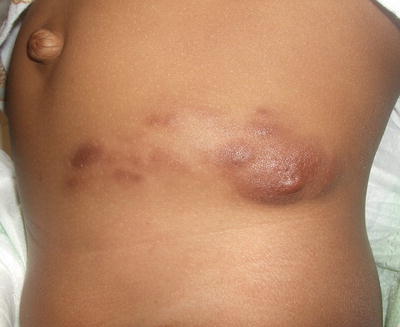
Fig. 25.13
Dermatomyofibroma presents as a firm, reddish brown nodular plaque extending across the left lower abdomen
25.4.2 Histology
Dermatomyofibroma appears as a plaque-like growth of spindle-shaped cells with uniform, long and tapered nuclei [55]. A pattern of intersecting fascicles is seen in some cases. The lesions are centered in the reticular dermis and may extend down into the superficial portion of the subcutaneous fat. In rare cases, lesions percolate deeper into the subcutis in a perpendicular orientation [56]. These neoplasms are variably cellular but have minimal cytologic atypia, and mitotic activity is quite rare. Thickened elastic tissue fibers and collagen fibers are interspersed between the slender nuclei [57].
Muscle actin is expressed by the tumor cells in some cases, which helps to differentiate this tumor from a cellular dermatofibroma, as does the horizontal orientation of the tumor cell population [57]. Dermatomyofibromas express smooth muscle actin in a minority of cases and do not express desmin. Calponin expression has also been described in these tumors [42]. Piloleiomyoma is another differential consideration. The fascicular growth pattern in piloleiomyomas is not present in dermatomyofibromas. Additionally, smooth muscle actin staining is uniform and intense in piloleiomyomas. A fibroblastic connective tissue nevus is also in the differential diagnosis, but the spindle-shaped cells in this entity express CD34 and not smooth muscle actin [58]. Electron microscopy shows that the tumor population consists of myofibroblasts [55].
25.4.3 Pathogenesis
The pathogenesis of dermatomyofibroma has yet to be determined, although it is known that the cellular component of this benign tumor has both fibroblastic and myofibroblastic differentiation [42].
25.5 Infantile Myofibromatosis
25.5.1 Clinical Features
Infantile myofibromatosis is a common fibrous tumor of infancy [59]. The exact incidence is unknown. Boys are more often affected than girls [60]. The majority of lesions appears within the first 2 years of life with 50–90 % appearing at birth or soon after [59, 60]. Three clinical variants exist. The most common variant is the solitary form, and it is characterized by a single firm, skin-colored or violaceous nodule [60]. Overlying telangiectasia or ulceration may occur. The multicentric variant is defined by involvement of the skin, subcutaneous tissues, muscles, and bones. The third variant is the generalized form with both cutaneous and visceral lesions .
Spontaneous regression is the rule in infantile myofibromatosis with involution over a 12- to 18-month period [60]. Local destruction secondary to mass effect can occur in the multicentric variant, although metastases are not seen. In the generalized variant, mortality rates approximate 75 % with death most often from complications due to cardiopulmonary or gastrointestinal involvement [59]. Surgical excision can be offered for symptomatic tumors without visceral compromise. The estimated risk of recurrence is 7–10 %. Other management options include radiotherapy, chemotherapy, and intralesional corticosteroids.
25.5.2 Histology
In infantile myofibromatosis, short fascicles of spindle-shaped cells form relatively sharply circumscribed nodules in the dermis. In some cases, there is a slightly infiltrative border. Collagen bundles surround these short fascicles. Necrosis and mitotic activity can be present, but are not abundant and atypical mitoses are not identified [61]. Some areas may resemble a hemangiopericytomatous growth pattern, and intravascular proliferation of the spindle-shaped cells has been reported. A bi-phasic growth pattern with a population of smaller, darker cells in the central portions of the lesions, and cells with more cytoplasm resembling smooth muscle located at the margins of the tumor is often described [62]. Monophasic variants also have been reported. In these cases, an abundance of small capillaries is recognized, raising the differential diagnosis of a primary vascular neoplasm [63]. Infantile myofibromatosis expresses smooth muscle actin, but it usually does not express desmin and myoglobin [64]. Ultrastructural examination reveals that the tumor cells have the appearance of myofibroblasts [65].
25.5.3 Pathogenesis
Infantile myofibromatosis is a benign fibrous tumor of childhood caused by abnormal proliferation of fibroblasts and myofibroblasts [66]. Mutations in platelet-derived growth factor receptor-beta (PDGFRB) , a receptor tyrosine kinase, is a cause of autosomal dominant infantile myofibromatosis. It has been shown that the mutations c.1681C>T (p.Arg561Cys) and c.1978C>A (p.Pro660Thr) in PDGFRB segregate with the disease in affected individuals [67, 68]. These mutations are activating mutations causing constitutive activation of the PDGF receptor.
Mutations in NOTCH3 have also been identified in infantile myofibromatosis [69]. NOTCH3 is a membrane protein involved in the NOTCH signaling pathway critical for embryonic development. Small molecule inhibitors of receptor tyrosine kinases that block PDGFRB and NOTCH3 may potentially be utilized to treat infantile myofibromatosis .
25.6 Fibroblastic Rheumatism
25.6.1 Clinical Features
Fibroblastic rheumatism is a rare disorder with approximately 30 reported cases to date, 31 % of which involve children [70]. Males are more commonly affected than females. The physical exam is characterized by tender, erythematous to skin-colored, small nodules usually on the hands and around the joints [70]. Additional features include sclerodactyly, joint contractures, thickened palmar fascia, and Raynaud’s phenomenon . Fibroblastic rheumatism also can be associated with an erosive arthropathy, which may predispose affected individuals to pronounced and persistent disability. A number of systemic therapies have been tried, including methotrexate with variable response [70].
25.6.2 Histology
Histologic sections of fibroblastic rheumatism reveal a poorly circumscribed dermal proliferation of spindle-shaped and stellate fibroblasts [71]. The proliferative cells have histologic and ultrastructural features of myofibroblasts [72]. The surrounded collagen bundles are markedly thickened, and there are diminished numbers of elastic tissue fibers within the proliferation. Surrounding fibrosis may be prominent [73]. A mild perivascular and interstitial lymphocytic infiltrate is observed in some cases [74, 75]. Immunohistochemical studies support the myofibroblastic differentiation in fibroblastic rheumatism with cells expressing smooth muscle actin [76]. Some authors have questioned whether this entity represents a variant of a non-Langerhans cell histiocytosis [77], while others believe it to be within the category of fibromatoses [74].
25.6.3 Pathogenesis
Fibroblastic rheumatism is a disorder with increased fibroblast proliferation. Histologic features suggest a myofibroblastic response to an unidentified stimulus [78, 79]. Transforming growth factor-β and granulocyte-macrophage colony-stimulating factor may be important in the development of this lesion, since they are known to induce the differentiation of fibroblasts into myofibroblasts [80].
25.7 Angiofibroma
25.7.1 Clinical Features
Multiple facial angiofibromas are commonly described as pathognomonic features of tuberous sclerosis and multiple endocrine neoplasia type 1 (Fig. 25.14) [81, 82]. However, there are other cutaneous variants of angiofibromas, including fibrous papule of the nose, pearly penile papules, and acral fibrokeratomas. Treatment may be warranted for cosmetic reasons as lesions are persistent [84].

Fig. 25.14
Angiofibromas appear as innumerable waxy, reddish brown papules primarily distributed along the central face in a patient with tuberous sclerosis
Angiofibromas are described as skin-colored to erythematous papules with clinical subtype dependent on the anatomic location. They are rarely symptomatic. Examples include the following:
- (a)
Fibrous papule—solitary, skin-colored papule typically on the nose of an adult.
- (b)
Pearly penile papule—shiny, skin-colored or white 1 mm papules along the corona of the penis in post-pubertal males [83].
- (c)
Periungual fibroma/Koenen tumor—a pathognomonic feature of tuberous sclerosis. This is skin-colored to pink papule typically found along the nail folds of multiple digits (Fig. 25.15) [82].
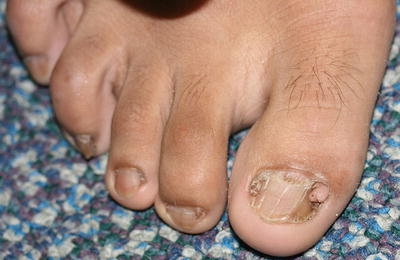
Fig. 25.15
Koenen tumors present as firm, skin-colored, filiform papules at the proximal nail fold of multiple toes in a teenage patient with tuberous sclerosis
25.7.2 Histology
The histologic changes in angiofibroma are identical to those seen in fibrous papules of the nose and pearly penile papules. It is likely that these entities are closely related, if not identical. The epidermis is either unremarkable or slightly flattened. The papillary dermis is characterized by dense fibrosis that obscures the boundary between papillary and reticular dermis (Fig. 25.16). Stellate and enlarged fibroblasts are present within the sclerotic collagen. Vessels within the dermis are enlarged (Fig. 25.17). “Onion skin” fibrosis is present as concentric rings around cutaneous appendages. Inflammatory, pleomorphic, and pigmented variants occur rarely [85]. A rare variant characterized by clear stellate fibroblasts has been reported in adults but not in children [86]. Other reported variants have granular stellate cells and epithelioid cells [87, 88].
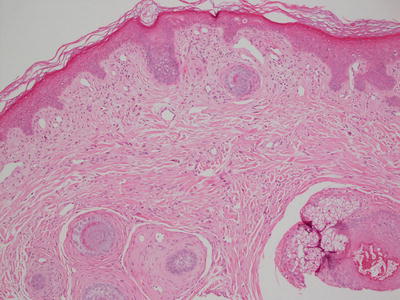
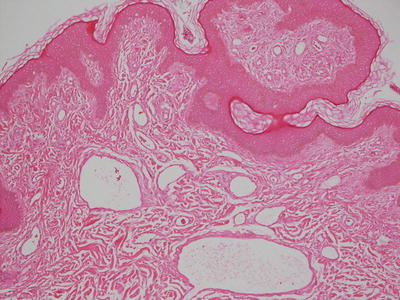
Fig. 25.16
Angiofibroma demonstrates collagenous overgrowth with concentric rings of fibrosis around hair follicles
Fig. 25.17
Vascular ectasia is a prominent finding in angiofibroma. Lesions occurring in patients with tuberous sclerosis are indistinguishable from sporadic ones
25.7.3 Pathogenesis
Angiofibroma is a cutaneous hamartoma of the face and nose that occurs sporadically and in genetic conditions such as tuberous sclerosis complex (TSC) . In TSC, angiofibroma presents as multiple lesions on the face before puberty [89, 90].
There is considerable overlap in the histologic features and pathogenesis between sporadic angiofibroma and angiofibroma associated with TSC. TSC is caused by germline mutation of either the TSC1 or TSC2 gene that encodes hamartin and tuberin, respectively [91, 92]. TSC1/TSC2 complex functions to inhibit the activation of the mammalian target of rapamycin (mTOR) signaling pathway. Loss of function of TSC complex leads to dysregulated activation of mTOR, and its downstream effectors p70 ribosomal protein S6 kinase (p70S6K) and ribosomal protein S6 (S6) [93, 94].
It has been proposed that keratinocytes in the epidermis and stromal cells in the dermis are the most likely cells that drive the formation of angiofibroma [95]. Stromal cells in angiofibroma express activated mTOR, p70S6K, and S6, which are effector molecules in the mTOR signaling pathway that cause TSC [91–93]. The proliferation of blood vessels, which is a feature in angiofibroma, may be induced by vascular endothelial growth factor (VEGF) produced by mTOR-activated cells [95]. Therapeutic intervention with mTOR inhibitors, such as topical rapamycin, has been successful in the treatment of angiofibroma in children and adults [96, 97].
The formation of angiofibroma in TSC patients may also be related to sun exposure and second-hit mutations. Studies of cells from skin tumors in TSC patients revealed germline and second-hit mutations in TSC1 and TSC2 genes [98]. Fifty percent of the somatic point mutations identified in TSC skin tumors were found exclusively in facial angiofibroma from sun-exposed sites. These results suggest that sun exposure may cause second-hit mutations and the development of TSC facial angiofibroma [98]. Avoidance of sun exposure may reduce the development of facial angiofibroma in children and adults with TSC.
25.8 Tuberous Sclerosis
25.8.1 Clinical Features
Tuberous sclerosis (TSC) is a hamartomatous disorder affecting the skin, brain, eyes kidneys, liver, heart, and lungs. Approximately 2 million people worldwide have tuberous sclerosis with an estimated prevalence of 1 in 6000–9000 individuals [99]. It is defined as an autosomal dominant disorder, although an estimated 50–65 % of affected individuals have a spontaneous mutation [99].
TSC is characterized by the triad of seizures, mental retardation, and facial angiofibromas, but only one third of affected individuals present with all of these features [82]. Asymmetrically distributed hypopigmented macules along the trunk and buttocks (an example being the “ash leaf macule”) appear by 2 years of age, and they are the initial cutaneous finding in many of the patients. Between 2 and 5 years of age, most patients develop angiofibromas at the central face, which are expansive especially following puberty [82]. Additional cutaneous features include fibrous forehead plaque, Shagreen’s patch at the lumbosacral cutaneous spine, periungal fibromas (Koenen tumors), molluscum fibrosum pendulum (soft pedunculated papules at the neck, axillae, and groin), and “confetti-like” hypopigmented macules at the distal extremities.
Phenotypic presentation and prognosis varies in TSC. However, the majority of patients will have some degree of epilepsy, often presenting in the first year of life [99]. Approximately fifty percent of affected individuals will have behavioral problems or autism spectrum disorders. Pulmonary lymphangiomyomatosis, which is primarily seen in women, and renal cell carcinoma, which has been reported in 2–4 % of cases, can be life threatening [99, 100].
25.8.2 Histology
Hypopigmented macules, Shagreen patches, forehead plaques, angiofibromas, and subungual fibromas are all seen with increased frequency in patients with TSC [101–103]. Café-au-lait macules are also present in patients with TSC [82].
The hypopigmented lesions show skin with unremarkable appearing epidermis and dermis. In contrast to the surrounding skin, the basal layer of the epidermis demonstrates decreased melanin within keratinocytes. The number of basilar melanocytes is normal. However, on ultrastructural examination, the melanocytes have fewer and smaller melanosomes. There appears to be decreased melanogenesis [104].
Shagreen patches are collagenomas or hamartomatous proliferations of collagen. Collagen bundles are increased in number and somewhat haphazard in their organization. The overgrowth of collagen bundles results in diminished number of elastic tissue fibers. The altered pattern of elastic tissue fibers is most easily detected on elastic tissue stains. The changes can be quite subtle, and comparison with adjacent tissue highlights the altered pattern.
Angiofibromas and subungual fibromas share similar histologic features with fibrous papules and pearly penile papules seen in individuals who do not have TSC. Subungual fibromas are uncommon in childhood [103]. The epidermis is flattened, and in some cases demonstrates slight melanocytic hyperplasia. The dermis shows increased and thickened collagen bundles that may be oriented perpendicular to the epidermis. Increased fibroblasts are present and may be somewhat enlarged. Concentric rings of collagen surround arterioles, venules, and capillaries that are somewhat dilated [105]. Forehead plaques demonstrate identical histologic features to those seen in angiofibroma and subungual fibroma [106].
25.8.3 Pathogenesis
Tuberous sclerosis complex is an autosomal dominant genetic disorder with an incidence of about 1 in 6000 [107]. The underlying pathogenesis of TSC is due to dysregulated intracellular signaling pathways that affect many cellular functions, including cell metabolism, survival and proliferation. TSC is caused by inactivating mutations of either the TSC1 or TSC2 tumor suppressor gene that encodes for the proteins hamartin (TSC1) and tuberin (TSC2) [91, 92]. Mutations in both TSC1 and TSC2 can produce the same phenotype with individual variations [108, 109]. Mutations in TSC2 are more common than mutations in TSC1, whether in familial cases (about 65 %) or in sporadic cases (about 75 %), and account for a ratio of TSC2:TSC1 mutations of about 2:1 to 3.5:1 [99]. Mutations in TSC2 produce more severe clinical manifestations with more hypomelanotic macules, more frequent neurologic and ophthalmologic symptoms, renal cysts, and perungual fibromas in male individuals [108].
TSC1/TSC2 complex is a tumor suppressor that negatively regulates the mammalian target of rapamycin (mTOR) signaling pathway, which is a serine/threonine kinase central to many cellular functions, including cell survival and metabolism [110–112]. mTOR is activated by the GTPase Rheb. Rheb in turn is negatively regulated by TSC1/TSC2 complex, which is a GTPase-activating protein that inactivates Rheb by stimulating the conversion of active Rheb-GTP to inactive Rheb-GDP. Loss of function of TSC1 or TSC2 leads to unregulated Rheb-GTP signaling and downstream mTORC1 activation. Constitutive activation of mTOR and its downstream effectors, including 4E-BP1 (eukaryotic translation initiation factor 4E-binding protein 1) and p70 S6-kinase, results in dysregulated cellular metabolic processes, protein synthesis and growth [113]. Given the pivotal role of mTOR signaling in the pathogenesis of TSC, mTOR inhibitors, such as rapamycin and its derivatives “rapalogs,” have been successfully used in the treatment of TSC [114, 115].
25.9 Sclerotic Fibroma
25.9.1 Clinical Features
Multiple sclerotic fibromas are commonly considered a feature of autosomal dominant hamartomatous genodermatoses, such as Cowden disease and tuberous sclerosis. However, isolated cases of solitary lesions have been described, most commonly in Caucasian adults [116]. Sclerotic fibromas are skin-colored to white, shiny to translucent papules with diameters between 2 and 9 mm, and may occur on any part of the body [116]. These lesions are benign but can be excised if desired.
25.9.2 Histology
The epidermis in sclerotic fibroma is flattened and overlies densely sclerotic collagen that has a “wood grain” appearance with alternating bands of white and dense eosinophilia (Figs. 25.18 and 25.19). The lesion is well circumscribed and displays minimal cellularity. Scattered fibroblasts are seen within the sclerotic dermis [116].
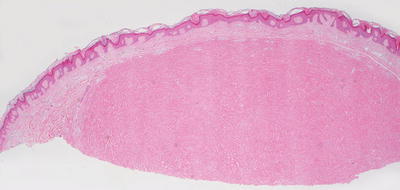
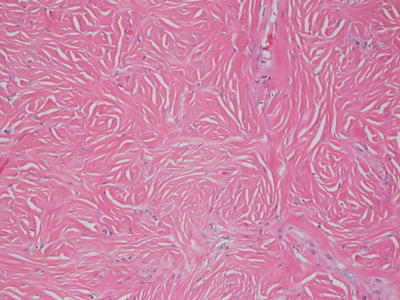
Fig. 25.18
Sclerotic fibroma demonstrates a well-circumscribed and sharply demarcated nodule consisting of thickened collagen bundles
Fig. 25.19
Areas with dense collagen and few nuclei give rise to a “wood grain” appearance in sclerotic fibroma
25.10 Infantile Digital Fibroma
25.10.1 Clinical Features
Infantile digital fibroma is a rare variant of superficial juvenile fibromatosis. The majority of lesions appear within the first 12 months of life, with one-third of cases present at birth [117].
Infantile digital fibromas present as skin-colored to erythematous papules and nodules along the dorsal and lateral aspects of the fingers or toes (Fig. 25.20) [117]. They are commonly multiple in number, affecting many digits and ranging in size to as large as 2 cm. Although benign with no known reports of local bony invasion or metastasis, larger lesions may cause joint deformities and contractures. Spontaneous regression appears to be the rule, making watchful waiting appropriate, especially since high recurrence rates are reported following excision.
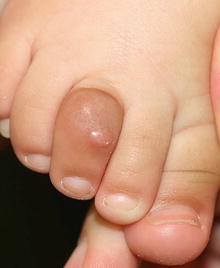
Fig. 25.20
Infantile digital fibroma appears as a shiny erythematous nodule on the dorsum of the right third toe in a young infant
25.10.2 Histology
In infantile digital fibroma, the epidermis demonstrates flattened rete ridges. Within the reticular dermis, there is a proliferation of plump myofibroblasts that have the characteristic peri-nuclear eosinophilic inclusions (Figs. 25.21 and 25.22) [118]. The inclusions are clumped actin filaments and stain strongly with anti-smooth muscle actin antibodies [119]. The nuclei are spindled with some stellate forms. In some cases, the fascicles of tumor cells and interspersed collagen may display a vertical orientation. Cytologic atypia is not present, and mitoses are rare. While based in the reticular dermis, the tumor is poorly circumscribed and may extend into the subcutis.
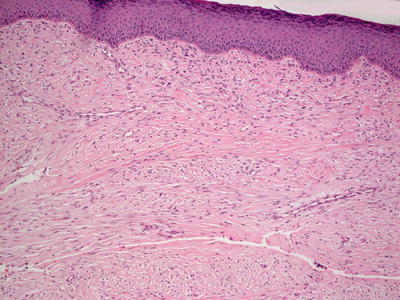
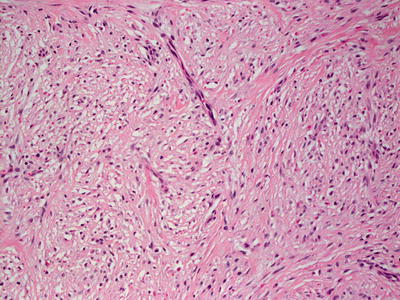
Fig. 25.21
Recurrent infantile digital fibroma is characterized by a dermis filled with fascicles of spindle-shaped cells with abundant eosinophilic cytoplasm
Fig. 25.22
Bland-appearing spindled cells demonstrate focal vacuolization and peri-nuclear inclusions in infantile digital fibroma
25.11 Acquired Digital Fibrokeratoma
25.11.1 Clinical Features
Acquired digital fibrokeratoma is rare and typically appears in middle-aged adults with a mean age of onset of 40 years of age [120]. Rare reports exist in individuals less than 18 years old.
Lesions most commonly occur on the fingers, but may be seen on other acral surfaces. They are skin-colored to pink, hyperkeratotic and exophytic papules, frequently with a surrounding collarette of skin. Acquired digital fibrokeratoma is usually solitary, but reports of multiple lesions have been described [120]. The lesion is a benign growth and may be removed by surgical excision.
25.11.2 Histology
Histologic changes in acquired digital fibrokeratoma include a normal or acanthotic epidermis overlying dense dermal collagen (Figs. 25.23 and 25.24). Hyperkeratosis is present as a secondary phenomenon due to trauma related to the exophytic nature of the lesion on an acral site. Markedly thickened collagen bundles are vertically oriented within the reticular dermis and extending into the papillary dermis (Fig. 25.24). Overgrowth of the collagen bundles results in loss of cutaneous appendages. Rare variants display increased numbers of fibroblasts and marked edema [121].
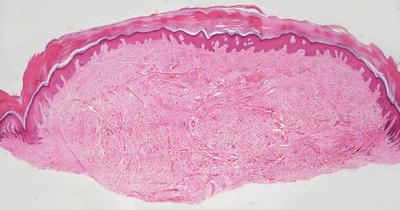
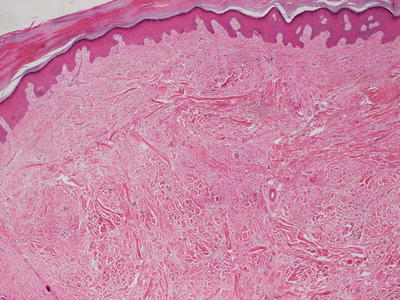
Fig. 25.23
Acquired digital fibrokeratoma demonstrates a polypoid growth on acral skin with collagenous overgrowth and lack of dermal appendages
Fig. 25.24
Collagen bundles are haphazardly arrayed and may be perpendicular to the skin surface in acquired digital fibrokeratoma
The differential diagnosis includes other dermal-based acral tumors, including supernumerary digits, subungual fibromas, neuromas, neurofibromas and sometimes plantar warts [122]. It is not clear whether peri-ungual fibrokeratomas represent the same tumor arising in a peri-ungual location [123, 124]. Supernumerary digits, neuromas, and neurofibromas display neural elements that are not present in acquired digital fibrokeratomas. In most cases, these structures are easily identified on routine sections, but special stains to identify neural elements may be helpful in some cases. Subungual fibromas do not have vertically oriented collagen, but often demonstrate peri-appendageal concentric fibrosis, stellate fibroblasts, and ectatic vessels. In some cases, the distinction from acquired digital fibrokeratoma may be difficult.
25.12 Acrochordon
25.12.1 Clinical Features
Acrochordons or “skin tags” are the most commonly seen fibrous proliferation. Prevalence increases with age, occurring equally in males and females [125]. Acrochordons are frequently seen in association with metabolic syndrome and in obese individuals [125].
Acrochordons present as skin-colored to hyperpigmented, pedunculated papules most commonly occurring at sites predisposed to persistent friction, such as the neck, axillae, and groin. Larger lesions with diameter up to 2 cm may be seen on the trunk. Acrochordons are benign but may be painful if they are irritated. Lesions can be removed easily by excision, cryosurgery, or electrodessication.
25.12.2 Histology
This common lesion has an epidermis that may be entirely normal, papillomatous, or acanthotic (Fig. 25.25). The dermis consists of variably ectatic blood vessels coursing in an unremarkable collagenous stroma. When there is trauma, the vessels may be markedly ectatic and thrombosed. Cutaneous appendages and adipocytes are ordinarily not present. Some authors use terms such as dermatolipoma for similar lesions that contain adipocytes, although there is little reason for this distinction because there is no difference in clinical course, prognosis or known systemic associations. Inflammation is present only secondary to trauma. Occasional stellate or pleomorphic fibroblasts may be present within the dermis in acrochordons. When they are extensive in number and mitoses are recognized, the name pseudosarcomatous polyp has been suggested, although these rare variants have not yet been described in children [126].
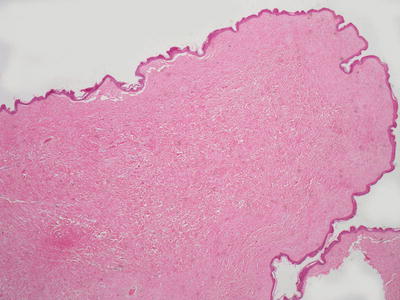
Fig. 25.25
Acrochordons are polypoid structures that generally lack cutaneous appendages and are composed largely of collagen
The differential diagnosis most commonly includes verruca vulgaris, although the characteristic epidermal changes of clumping of keratohyalin granules, focal parakeratosis, and keratinocyte vacuolization are not seen in acrochordons .
25.12.3 Pathogenesis
Acrochordon is often linked to type 2 diabetes mellitus and obesity. Acrochordon is also associated with hereditary autosomal dominant Birt–Hogg–Dube syndrome, which is characterized by multiple fibrofolliculomas, trichodiscomas, and acrochordons [127]. Increased levels of tumor necrosis factor-α (TNF-α), which is produced by mast cells and macrophages, have been found in acrochordon as compared with normal skin [128]. Some studies have suggested the role of local trauma in the pathogenesis of acrochordon through possible trauma-induced upregulation of TRAIL and induction of mast cells migration into the site of trauma [128]. TNF-α released by mast cells may result in changes leading to acrochordon formation .
25.13 Angiofibroblastoma
25.13.1 Clinical Features
Angiofibroblastoma is a newly described rare fibrous growth occurring on the extremities. The exact prevalence is unknown with no documented gender predilection [129]. Lesions are characterized as solitary, non-descript subcutaneous nodules typically presenting on the extremities. Angiofibroblastomas have a benign clinical course and respond well to simple excision with no documented evidence of recurrence or metastasis.
25.13.2 Histology
This rare neoplasm is characterized by a dermal proliferation of fibroblasts coursing in a myxoid stroma. The epidermis is uninvolved. The fibroblasts are spindled with abundant stellate-shaped cells. Mitotic activity is rare, and there is no significant cytologic atypia in the fibroblastic proliferation. In some areas, the stroma is comprised of dense collagen instead of the myxoid stroma seen in the majority of the lesions. Many small capillaries are present admixed throughout the myxoid stroma and account for the naming of this entity [129, 130].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


