12 Cytomegalovirus, Epstein–Barr virus and BK virus infection following solid-organ transplantation
Cytomegalovirus
 Cytomegalovirus (CMV) infection remains a significant contributor to post-transplant morbidity and mortality. Even asymptomatic CMV infection is associated with a 2.9 hazard ratio of death in renal transplant recipients by 4 years after transplant.1
Cytomegalovirus (CMV) infection remains a significant contributor to post-transplant morbidity and mortality. Even asymptomatic CMV infection is associated with a 2.9 hazard ratio of death in renal transplant recipients by 4 years after transplant.1
Once active infection has been established, CMV replication is highly dynamic, with rapid increases in viral loads, development of CMV viraemia and an elevated risk of invasive disease. Exposure to the virus, as indicated by the presence of detectable IgG anti-CMV antibodies in the plasma, increases with age in the general population and is present in more than two-thirds of donors and recipients prior to transplantation. The virus can be transmitted from blood transfusion or by the transplanted kidney. The administration of immunosuppressive drugs, especially lymphocyte depleting, increases the risk of clinically relevant disease.2
CMV infection versus disease
There is an important distinction between CMV infection and disease. Infection is active if one or more of the following findings is noted: seroconversion with the appearance of anti-CMV IgM antibodies; a fourfold increase in pre-existing anti-CMV IgG titres; detection of CMV antigenaemia; detection of CMV DNAaemia by molecular techniques; or isolation of the virus by culture. CMV disease, in comparison, requires clinical signs and symptoms, such as fever, leukopenia or organ involvement.3 In general terms, the total burden of CMV viral particles in the host correlates with clinical evidence of disease, disease severity or response to therapy.4,5
CMV disease
The most common presentation of CMV disease is a mononucleosis-like syndrome with fever, malaise, myalgias and arthralgias, usually with leucopenia and mild (5–10%) atypical lymphocytosis. One of the cardinal features of CMV is leucopenia. This is more common in the absence of maintenance steroids, which are increasingly popular. A mild elevation in serum aminotransferase concentrations may also occur. With the use of mycophenolate mofetil (MMF), invasive CMV disease can occur in the absence of fever and leucopenia.
Interstitial pneumonitis and ulcerations in the oesophagus and colon cause major morbidity. For example, gastrointestinal bleeding among renal transplant recipients is commonly caused by erosions due to CMV. In this setting, invasive disease is usually confirmed with endoscopic biopsy and may be present in the absence of viraemia. By comparison, encephalopathy and chorioretinitis are unusual in renal transplant recipients but also may be present in the absence of viraemia (Box 12.1).
Treatment of CMV disease
Patients with organ involvement may benefit from a 2- to 3-week course of intravenous ganciclovir.7 Hyperimmune globulin (Cytogam) may also be added to the treatment regimen in those with organ involvement.8
The usual dose of ganciclovir is 5 mg/kg i.v. every 12 hours in patients with normal renal allograft function. Although dose reductions are recommended for renal insufficiency, lower doses increase the likelihood of recurrent CMV disease in renal transplant recipients.7
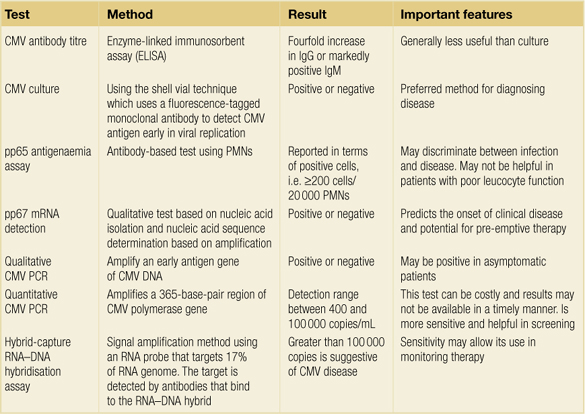
 The effectiveness of valganciclovir was best evaluated in the VICTOR study.9 In this non- inferiority trial, 321 solid-organ recipients with CMV disease were randomly assigned to oral valganciclovir (900 mg twice daily for 21 days) or intravenous ganciclovir (5 mg/kg twice daily for 21 days), which was followed in both arms by valganciclovir (900 mg daily until day 49). Both agents were similarly effective in suppressing viraemia at 21 days.
The effectiveness of valganciclovir was best evaluated in the VICTOR study.9 In this non- inferiority trial, 321 solid-organ recipients with CMV disease were randomly assigned to oral valganciclovir (900 mg twice daily for 21 days) or intravenous ganciclovir (5 mg/kg twice daily for 21 days), which was followed in both arms by valganciclovir (900 mg daily until day 49). Both agents were similarly effective in suppressing viraemia at 21 days.
CMV-induced renal disease
Whether the virus itself can cause allograft dysfunction is unclear.10 Renal function may deteriorate in patients with CMV infection but factors such as decreased renal perfusion, acute tubular necrosis and transplant rejection may be more important than a direct viral effect on the kidney. CMV infection has been identified as an independent risk factor for the development of rejection.11 Infection with CMV has also been implicated in the development of coronary artery narrowing.12
CMV has also been associated with thrombotic microangiopathy (HUS/TTP) in solid-organ and bone marrow transplant recipients, which may respond to immunoglobulin infusion.13–15
Prevention
Currently, there are two principal approaches to prevent CMV disease in patients undergoing solid-organ transplantation. A prophylactic strategy with the administration of antiviral agents to patients at increased risk of developing CMV infection and a pre-emptive strategy with periodic monitoring for viraemia, principally using polymerase chain reaction (PCR), to permit treatment of asymptomatic early systemic infection. It is unclear which strategy is preferred. A number of meta-analyses of these and other strategies have been performed.16–18
A 2006 systematic review evaluated 10 trials involving 476 solid-organ transplant (SOT) recipients, of which six were pre-emptive therapy versus placebo or standard therapy (treatment once CMV disease occurred), three were pre-emptive versus prophylactic therapy, and one was oral versus intravenous pre-emptive therapy.16 No difference was observed with pre-emptive versus prophylaxis therapy in the risks of CMV disease or mortality. Oral and intravenous pre-emptive therapy were also associated with similar risks of disease.
A 2006 meta-analysis of 17 trials (with the comparator being placebo or no treatment) involving 1980 kidney and liver transplant recipients found that CMV organ disease was significantly reduced with both prophylaxis and pre-emptive strategies.17 Acute rejection was significantly reduced with both. Only prophylaxis resulted in decreased bacterial and fungal infections and mortality.
Prophylactic therapy
A meta-analysis of 19 trials in solid-organ transplant recipients found that antiviral prophylaxis significantly lowered the risk of CMV disease, CMV infection and mortality.18 Ganciclovir was superior to acyclovir, while oral ganciclovir was similarly effective as valganciclovir and intravenous ganciclovir.
Ganciclovir and valganciclovir
Ganciclovir or valganciclovir has now supplanted either acyclovir or CMV hyperimmune globulin as the prophylactic therapy of choice for CMV infection or disease among transplant recipients. Although prophylactic intravenous ganciclovir can prevent CMV infection in transplant recipients it is unnecessary, expensive and associated with neutropenia, and oral ganciclovir or valganciclovir have taken over as prophylaxis. Although oral ganciclovir has poor bioavailability, one study, which evaluated 42 renal transplant recipients at risk for CMV infection in whom induction therapy was administered, found that prophylaxis with ganciclovir (1000 mg t.i.d.) was superior to deferred therapy with acyclovir (200 mg b.i.d.), both given for 12 weeks.19
Another prospective study of 101 high-risk renal transplant recipients, who were randomly assigned to prophylactic therapy with oral ganciclovir or acyclovir, found significantly fewer CMV infections after 3 months of therapy with ganciclovir.20 A larger trial of liver transplant recipients (none of whom were seronegative recipients of seronegative donors) were randomised to either 1000 mg t.i.d. of oral ganciclovir or placebo. At 6 months, active therapy significantly reduced the incidence of CMV disease.21
Valganciclovir, a valyl-ester prodrug of oral ganciclovir, is rapidly metabolised to an active form (ganciclovir) in the intestinal wall and liver. It has a bioavailability of nearly 70% (compared to 7% for oral ganciclovir) and at doses of 450–900 mg produces serum ganciclovir levels that are similar to that measured with intravenous administration of ganciclovir administered at 2.5–5 mg/kg.22
 In the phase III PV16000 international, double-dummy, double-blinded trial of oral valganciclovir versus oral ganciclovir for the prevention of CMV, valganciclovir and oral ganciclovir had similar efficacy and reaffirmed that a period of intravenous ganciclovir is unnecessary when an appropriate oral agent is used. During prophylaxis only 8.0% and 1.6% respectively developed symptomatic CMV infection; CMV disesase was common after prophylaxis, with disease occurring in 23% and 22% respectively by 6 months. These results suggest that a duration for CMV prophylaxis of 100 days may be insufficient for CMV D + R transplant recipients23 (Fig. 12.1).
In the phase III PV16000 international, double-dummy, double-blinded trial of oral valganciclovir versus oral ganciclovir for the prevention of CMV, valganciclovir and oral ganciclovir had similar efficacy and reaffirmed that a period of intravenous ganciclovir is unnecessary when an appropriate oral agent is used. During prophylaxis only 8.0% and 1.6% respectively developed symptomatic CMV infection; CMV disesase was common after prophylaxis, with disease occurring in 23% and 22% respectively by 6 months. These results suggest that a duration for CMV prophylaxis of 100 days may be insufficient for CMV D + R transplant recipients23 (Fig. 12.1).
Valacyclovir may prevent CMV disease among high-risk renal transplant patients. In a multicentre prospective study, use of valacyclovir was associated with a reduced incidence of disease among both seronegative and seropositive recipients.24 Valacyclovir was also associated with a reduced incidence of acute rejection among seronegative patients (26% vs. 52% vs. placebo, P = 0.001). Since valacyclovir (even in high concentrations) has limited activity against CMV, the mechanism of action for these results remains unclear.
CMV hyperimmune globulin
A multicentre trial, performed before the availability of ganciclovir, showed that hyperimmune globulin decreased the incidence of CMV disease (20% vs. 60%), reduced the incidence of fungal or parasitic superinfection (0% vs. 20%) and diminished the incidence of marked leucopenia, a marker of severe infection (4% vs. 37%), but did not alter the incidence of viral isolation or seroconversion.25
Other antiviral agents
Leflunomide
Leflunomide is a pyrimidine synthesis inhibitor and immunosuppressive agent currently utilised for the treatment of rheumatoid arthritis. Given its immunosuppressive properties, it has also been used in experimental and clinical transplantation. Leflunomide prevents CMV and herpes simplex virus (HSV) type I replication by interfering with virion assembly, and may have a role in the management of CMV in transplant recipients.26–30
Acyclovir
 The efficacy of acyclovir as prophylaxis in CMV infection has been compared to other antiviral agents, particularly ganciclovir. A 3-month course of high-dose acyclovir (800 mg four times per day, with modification of dosage for impaired renal allograft function) after transplantation has also been utilised. In one randomised, placebo- controlled trial, symptomatic CMV disease developed in only one of six CMV-negative recipients who received a graft from a CMV- positive donor versus all seven patients treated with placebo.31 More recent studies have shown that acyclovir has minimal utility for prophylaxis for CMV.32
The efficacy of acyclovir as prophylaxis in CMV infection has been compared to other antiviral agents, particularly ganciclovir. A 3-month course of high-dose acyclovir (800 mg four times per day, with modification of dosage for impaired renal allograft function) after transplantation has also been utilised. In one randomised, placebo- controlled trial, symptomatic CMV disease developed in only one of six CMV-negative recipients who received a graft from a CMV- positive donor versus all seven patients treated with placebo.31 More recent studies have shown that acyclovir has minimal utility for prophylaxis for CMV.32
Maribavir
Maribavir is a benzimidazole nucleoside that prevents viral DNA synthesis and capsid nuclear egress. It is therefore useful for CMV infections resistant to ganciclovir, cidofovir or foscarnet. In July 2004, a phase II trial was initiated.33
Drug-resistant CMV
Ganciclovir-resistant CMV isolates have emerged among seronegative recipients of seropositive organs. In one study of 240 recipients of liver, kidney or pancreas transplants, ganciclovir-resistant CMV disease developed in 7% of seronegative recipients of CMV-positive organs compared to none of 173 seropositive recipients.34 In the PV16000 trial, three cases (2%) of those administered oral ganciclovir developed ganciclovir resistance compared to none of the valganciclovir patients.23
CMV status and prophylactic therapy
CMV-positive donor, CMV-negative recipient (D+R−)
Historically, approximately 70–90% of CMV- positive donor, CMV-negative recipients developed ‘primary’ CMV infection when prophylaxis was not administered. Furthermore, 50–80% developed CMV disease and roughly 30% developed pneumonitis, a major contributor to morbidity and mortality and, in the absence of prophylactic therapy, the mortality rate was 15%. For unclear reasons, the rate of primary infection has decreased to 50% even in the absence of prophylaxis and despite the use of lymphocyte-depleting therapy.35–37
CMV-negative donor, CMV-positive recipient (D−R+)
Seropositive recipients of seronegative organs may have reactivation of latent CMV infection due to the administration of immunosuppressive drugs. CMV infection or disease may develop in up to 20% of cases, although progression to pneumonitis is rare. Some groups use prophylactic ganciclovir therapy in this population. This is especially true when lymphocyte-depleting agents are used for induction therapy or treatment of rejection.36
CMV-positive donor, CMV-positive recipient (D+R+)
Seropositive recipients of CMV-positive kidneys are at risk for reactivation of latent virus or superinfection with a new viral strain. The worst graft and patient survival at 3 years post-transplantation is observed among those in which the donor and recipient are both positive.38,39 This unexplained increased risk of disease may be due to the presence of multiple CMV virotypes or reactivation of different viruses.
CMV-negative donor, CMV-negative recipient (D−R−)
The incidence of disease in the D−R− population is 5–10%. Given this low prevalence of disease, independent of type of immunosuppressive therapy, no treatment with anti-CMV prophylaxis is recommended.36
Duration of prophylaxis
At some transplant centres, a longer duration of prophylaxis has been used according to the CMV serostatus of the donor and recipient.35,40,41 In one centre, prophylaxis with oral ganciclovir was prescribed for 90 days in D−R+ cases and for 180 days in D+R− and D+R+ cases.35 There was a significant association of disease with donor CMV seropositivity when the recipient was CMV seronegative. By comparison, donor CMV serostatus was not significantly associated with CMV disease when the recipient was CMV seropositive. All CMV disease occurred after prophylaxis ended. Despite the relatively mild severity of disease, which was promptly detected and treated, the presence of disease was significantly associated with poorer graft survival.
In another study, the group in which recipients received a 24-week regimen of oral ganciclovir, compared with a historic control group administered a 12-week course, a markedly lower rate of CMV infection occurred (31% vs. 7% respectively).40 A similar decrease in CMV was found with a 6-month course of valganciclovir.41
Pre-emptive strategy
A recent systematic review showed that pre- emptive therapy, compared with placebo or standard therapy, significantly reduced the risk of CMV disease.42
 In a single-centre study comparing pre-emptive and prophylactic strategies, 98 at-risk kidney transplant recipients (D+R−, D+R+, D−R+) were randomly assigned to prophylaxis (valganciclovir, 900 mg/day for 100 days) or pre-emptive therapy (valganciclovir, 900 mg twice per day for at least 21 days for CMV DNAaemia as defined by levels greater than 2000 copies/mL by PCR). All patients who developed CMV DNAaemia were treated with valganciclovir, which was continued until the PCR assay became negative.37 A similar incidence of symptomatic infection (disease) was reported in both arms (overall 4%) and there were no deaths. CMV DNAaemia was significantly more likely in the pre-emptive group (59% vs. 29% for the prophylactic arm). However, the late onset of CMV DNAaemia (greater than 100 days) was significantly more likely with prophylaxis therapy (24% vs. 0%). Time of onset of disease occurred later than 100 days in all patients with symptomatic disease in the prophylactic arm, but at day 62 in one patient treated pre-emptively. Both approaches were associated with similar overall costs, minimal side-effects and low rates of adverse outcomes.
In a single-centre study comparing pre-emptive and prophylactic strategies, 98 at-risk kidney transplant recipients (D+R−, D+R+, D−R+) were randomly assigned to prophylaxis (valganciclovir, 900 mg/day for 100 days) or pre-emptive therapy (valganciclovir, 900 mg twice per day for at least 21 days for CMV DNAaemia as defined by levels greater than 2000 copies/mL by PCR). All patients who developed CMV DNAaemia were treated with valganciclovir, which was continued until the PCR assay became negative.37 A similar incidence of symptomatic infection (disease) was reported in both arms (overall 4%) and there were no deaths. CMV DNAaemia was significantly more likely in the pre-emptive group (59% vs. 29% for the prophylactic arm). However, the late onset of CMV DNAaemia (greater than 100 days) was significantly more likely with prophylaxis therapy (24% vs. 0%). Time of onset of disease occurred later than 100 days in all patients with symptomatic disease in the prophylactic arm, but at day 62 in one patient treated pre-emptively. Both approaches were associated with similar overall costs, minimal side-effects and low rates of adverse outcomes.
Recommendations
With a prophylactic strategy, oral ganciclovir or valganciclovir should be administered to all patients other than seronegative recipients of seronegative grafts for a total duration of 100 days.3 The choice of agent depends upon the renal function of the patient and the relative affordability of the agents. In general, valganciclovir is preferred because of the reduced pill burden, increased likelihood of compliance with daily administration, and the potential for a decreased incidence of drug resistance. However, valganciclovir is contraindicated when the patient is on dialysis or has an estimated creatinine clearance less than 10 mL/min.
Epstein–Barr virus
Epstein–Barr virus (EBV) is a widely disseminated herpes virus spread by intimate contact between susceptible persons and asymptomatic EBV shedders. The majority of primary EBV infections throughout the world are subclinical. Approximately 90–95% of adults are EBV seropositive worldwide.6
Virology
EBV is a member of the gamma herpes virus family and the prototype for the lymphocryptovirus group of viruses that typically result in latent infection with persistence of the viral genome and expression of a restricted set of latent gene products. Host cells in humans are B lymphocytes, T lymphocytes, epithelial cells and myocytes. Unlike herpes simplex (HSV) or CMV, EBV is capable of transforming B cells and does not routinely display a cytopathic effect in cell culture.
Viral receptor
The EBV receptor on human cells is the B-cell- surface molecule CD21, which is the receptor for the C3d component of complement (also called CR2, complement receptor type 2).43 Infection is initiated by binding of the major EBV outer envelope glycoprotein gp350/220 with CD21.
The virus is then transported to the nucleus and the EBV genome is replicated by cellular DNA polymerases during the cell cycle S phase.44 It persists as multiple, extrachromosomal double-stranded EBV episomes, which are organised into nucleosomes similar to chromosomal DNA.45
Virus expression in latent infection
EBV exploits normal pathways of B-cell differentiation to allow it to persist in a transcriptionaly quiescent state in memory B cells and thus minimise immune recognition.46,47 Intracellular persistence of the entire viral genome is achieved through circularisation of the linear EBV genome, and maintenance of multiple copies of this covalently closed episomal DNA.44
Clinical manifestations
Primary infection
Primary EBV infection in infants and children
Primary EBV infections in infants and young children are common and frequently asymptomatic.48 When symptoms occur, a variety of manifestations have been observed, including otitis media, diarrhoea, abdominal complaints, upper respiratory infection and infectious mononucleosis. Children can have symptomatic primary EBV infection without the production of heterophile antibodies which are insensitive for diagnosis. Thus, EBV-specific serological studies are required to establish the diagnosis definitively. However, antiviral capsid antigen (anti-VCA) IgM was less frequently positive in infants; peak titres of VCA antibody were lower, and the development of antibodies to early antigen was less common in infants.
Acute infectious mononucleosis
Infectious mononucleosis is the best-known acute clinical manifestation of EBV. It often begins with malaise, headache, low-grade fever before development of the more specific signs of tonsillitis or pharyngitis, cervical lymph node enlargement and tenderness, and moderate to high fever.48,49 Affected patients usually have peripheral blood lymphocytosis, composed of atypical lymphocytes. The lymphadenopathy is characteristically symmetric and involves the posterior cervical chain more than the anterior chain. Tonsilar exudates are a frequent component of the pharyngitis, and can be white, grey–green or necrotic in appearance. Splenomegaly occurs in up to 50%, but jaundice and hepatomegaly are uncommon. Acute symptoms resolve in 1–2 weeks, but fatigue often persists for months. The vast majority of individuals with primary EBV infection recover uneventfully and develop a high degree of durable immunity.
Other manifestations
Neurological syndromes include Guillain–Barré syndrome, facial nerve palsy, meningoencephalitis, aseptic meningitis, transverse myelitis, peripheral neuritis and optic neuritis.50 Haematological abnormalities include haemolytic anaemia, thrombocytopenia, aplastic anaemia, thrombotic thrombocytopenic purpura/haemolytic–uraemic syndrome, and disseminated intravascular coagulation. EBV can affect virtually any organ system and has been associated with pneumonia, myocarditis, pancreatitis, mesenteric adenitis, myositis, glomerulonephritis and genital ulceration.51
Complications
EBV infection is associated with a number of acute complications, including a morbilliform rash following the administration of ampicillin and, to a lesser extent, penicillin. Oral hairy leucoplakia (OHL) is an unusual EBV-mediated mucocutaneous disease of the lingual squamous epithelium.52 The OHL lesions appear to be relatively specific for human immunodeficiency virus (HIV) infection, since they are only rarely observed in patients with other immunodeficiencies.52,53 The use of highly active antiretroviral therapy has reduced the incidence of OHL.54Splenic rupture is a rare but potentially life-threatening complication of infectious mononucleosis, estimated to occur in between one and two cases per 1000.55Obstruction of the upper airway due to massive lymphoid hyperplasia and mucosal oedema is an uncommon and potentially fatal complication of infectious mononucleosis.
Lymphoproliferative disorders
EBV infection is associated with a variety of lymphoproliferative disorders, including haemophagocytic lymphohistiocytosis, lymphomatoid granulomatosis, X-linked lymphoproliferative disease (also called Duncan syndrome) and EBV-associated post-transplant lymphoproliferative disease (PTLD).56 EBV is a transforming virus and has also been causally linked to a variety of malignancies in addition to lymphomas in transplant recipients.56 These include Burkitt’s lymphoma (tumours in HIV-infected patients), Hodgkin lymphoma, nasopharyngeal and other head and neck carcinomas, and T-cell lymphoma.
Treatment and prevention
Anecdotal use of interleukin-2, interferon-α and intravenous immunoglobulins have been reported. No clear benefits of such modalities have been demonstrated, with the possible exceptions of lymphomatoid granulomatosis and PTLD.57
The large body of evidence implicating EBV in the aetiology of a variety of human neoplasms has made the prospect of developing a viral-based vaccine effective against human cancers appealing. The viral glycoprotein gp350/220 is the most abundant capsid protein present in lytically infected cell plasma membranes and the most abundant protein on the outer surface of the virus. It binds to the CD21 receptor on the B cell, and is responsible for the initiation of infection. Most of the human EBV-neutralising antibody response is directed against gp350/220.58,59 Thus, gp350/220 is the major EBV lytic-cycle gene product being pursued in the development of a subunit vaccine. In animal studies, immunisation with partially purified gp350/220 antigen induced EBV-neutralising antibody and protected a portion of cotton-top tamarins against a normally lethal, lymphoma-producing challenge with EBV.60
Post-transplant lymphoproliferative disorder (PTLD)
Lymphoproliferative disorders occurring after transplantation have different characteristics from those that occur in the general population.61 Non-Hodgkin’s lymphoma (NHL) accounts for 65% of lymphomas in the general population compared to 93% in transplant recipients. These tumours are mostly large-cell lymphomas, the great majority of which are of the B-cell type.61 Extranodal involvement is common, occurring in approximately 30–70% of cases.
Although uncommon, PTLD may also originate from T cells and more rarely from natural killer cells.62–66 As of 2004, only 17 cases of T-cell PTLD had been reported among renal transplant recipients.66
Pathogenesis
The pathogenesis of post-transplant NHL in most patients is related to B-cell proliferation induced by infection with EBV in the setting of chronic immunosuppression.56 However, EBV-negative disease can occur.67
Most PTLD cells in solid-organ allograft recipients are of host origin. The clinical manifestations, and course of PTLD, vary with the origin of the lymphoproliferative cells.68–70 In a report of 12 renal transplant recipients, eight arose from the recipient and four from the donors.69 Recipient-origin PTLD presented as multisystem disease at a mean of 76 months after transplantation; five of the eight patients with disease from the recipient died. In contrast, donor-origin PTLD was limited to the allograft, developed after a mean of 5 months after transplantation, and regressed after reduction of immunosuppression.
Clinical manifestations and epidemiology
Three types of EBV-related lymphoproliferative disease occur in transplant recipients.71 Benign polyclonal lymphoproliferation is an infectious mononucleosis-type acute illness that develops 2–8 weeks after immunosuppressive therapy begins and accounts for 55% of cases.71 It is characterised by polyclonal B-cell proliferation with normal cytogenetics and no evidence of immunoglobulin gene rearrangements to suggest malignant transformation. The second EBV-induced disorder is similar to the first in its clinical presentation, but is characterised by polyclonal B-cell proliferation with evidence of early malignant transformation, such as clonal cytogenetic abnormalities and immunoglobulin gene rearrangements and accounts for approximately 30% of cases.
The third type is characterised by monoclonal B-cell proliferation with malignant cytogenetic abnormalities and immunoglobulin gene rearrangements, and accounts for about 15% of cases. It is usually an extranodal condition presenting with localised solid tumours.71 Involved organs include the gastrointestinal tract (stomach, intestine), lungs, skin, liver, central nervous system (CNS) and the allograft itself; 20–25% have CNS disease (which is rare in the general population) and a similar proportion have infiltrative lesions in the allograft.72
Non-EBV-associated PTLD differs clinically from EBV-related tumours.67,73,74 Tumours not due to EBV present much later, suggesting that their incidence may increase with time, and are much more aggressive.
The overall incidence of PTLD is approximately 1%, 30–50 times higher than in the general population, with a recent trend towards increased frequency.75 The risk of PTLD is greatest in patients with more marked degrees of immunosuppression, which explains part of the variability in the incidence of PTLD with different types of transplants.76 The incidence is 1–2% in liver transplants, 1–3% in renal transplants, 2–6% in heart transplants, 2–9% in lung transplants and as high as 11–33% in intestinal or multiorgan transplants.77–86
Risk factors
The principal risk factors underlying the development of PTLD are the degree of overall immunosuppression and the EBV serostatus of the recipient. Additional risk factors include time post-transplant, recipient age and ethnicity.75,87
Degree of immunosuppression
 Solid-organ transplant patients at increased risk for PTLD are paediatric recipients and those treated with an increased degree of immunosuppression, particularly those exposed to certain types of lymphocyte-depleting therapy.79,80,83,88–90
Solid-organ transplant patients at increased risk for PTLD are paediatric recipients and those treated with an increased degree of immunosuppression, particularly those exposed to certain types of lymphocyte-depleting therapy.79,80,83,88–90
The incidence ratio of PTLD, compared with the non-transplant population, was 21.5, 4.9, 29.0, 21.6 and 7.8 for those administered induction therapy with OKT3, antithymocyte globulin (ATG), ATGAM, thymoglobulin and interleukin-2 receptor antagonists respectively.90
For OKT3-treated patients, both the dose and the duration of therapy are important. The incidence was 11% in those treated with OKT3 for induction and 36% (5 of 14) in patients who received more than 75 mg of OKT3.83 OKT3 is not currently used commonly for induction therapy.
The incidence of PTLD is highest in the first year and falls by about 80% thereafter. The incidence of PTLD is much greater in heart transplant recipients in whom a greater degree of immunosuppression is required because of the more serious consequences of transplant rejection. PTLD tends to be ‘organotropic’. Renal transplant recipients are more likely to have renal lymphoma, while heart transplant recipients are more likely to develop lymphoma in the heart or lungs.79 In a report of nine lung transplant recipients with PTLD, eight had isolated intrathoracic disease.91 Local immune reaction against the graft may be one of the factors promoting malignant transformation.
The risk of PTLD is also significantly higher among those administered tacrolimus versus ciclosporin for maintenance therapy without induction therapy.88,89 If induction therapy were given, there was a non-significant trend toward a higher risk with tacrolimus.88 The use of MMF appears not be associated with an increased risk of PTLD.92
EBV serostatus
EBV serostatus is considered an important risk factor for development of PTLD. In the absence of the use of OKT3 and CMV seromismatch (i.e. a negative recipient and a positive donor), the incidence rate of PTLD for EBV-seronegative recipients was 24 times higher than that for EBV-seropositive recipients.93 These patients, who had no preoperative immunity to EBV, usually acquire the infection post-transplant from the donor.93 PTLD developed in 42% of patients who developed primary EBV infection after transplantation.84,94
Other risks
Additional risk factors for PTLD include a history of pretransplant malignancy and fewer HLA matches.95,96
PTLD is also more common in children because more are EBV seronegative prior to transplantation. A prospective study of 50 paediatric heart transplant recipients found that the overall incidence of PTLD was 26%.97 However, the incidence varied according to the initial and final EBV status, occurring in 12 of 19 (63%) who seroconverted after transplantation, 1 of 20 (5%) who were seropositive before transplantation, and 0 of 12 who were initially seronegative and remained so after transplantation.
The risk of PTLD also varies with time post- transplant. In a retrospective analysis of nearly 90 000 patients placed on the renal transplant waiting list over a 10-year period, 357 cases of lymphoma developed in transplant recipients.87 The first year post-transplant was associated with the highest lymphoma rate. An increased risk was also noted in those under 25 years of age and Caucasian.
A combination of risk factors has been shown to markedly increase the overall risk of PTLD in non-renal transplant recipients.93 Therapy with OKT3 for rejection and CMV sero-mismatch increases the risk four- to sixfold above that seen in EBV- seronegative recipients. The presence of all three risk factors increased the incidence rate of fatal and/or CNS PTLD by a factor of 654 compared to patients lacking all three factors.
Evaluation and diagnosis
An accurate diagnosis of PTLD requires a high index of suspicion, since the disorder may present subtly and/or extranodally.98 Quantitative viral loads exceeding 1000 copies/mL in plasma or 5000 copies/mL are suggestive but not specific with a poor predictive value.99 Radiological evidence of a mass or the presence of elevated serum markers (such as increased LDH levels) are suggestive of PTLD, with positive positron emission tomography (PET) scanning (possibly indicating metabolically active areas) also favouring the diagnosis.100 PET/computed tomography (CT) is a useful tool for staging and therapy monitoring of PTLD after liver transplantation.101
The different forms of PTLD are diagnosed histologically and distinguished by a number of features. These include clonality, whether elements of a malignant process are present (such as the presence or absence of clonal cytogenetic abnormalities, immunoglobulin gene rearrangements, and disruption of underlying tissue architecture), donor versus recipient origin, and (increasingly) whether EBV can be detected within the tumour.76 A number of classification schemes have been published.102 The reliance upon histological descriptions of PTLD is limited because of the lack of uniformity concerning definitions of a polyclonal or monoclonal process, diagnostic use of non-histological features, such as DNA rearrangements, mutations and clonality, use of EBV positivity within the tumour or the pathological process, and donor or host origin of the tumour.
Confirming the presence of cardiac lymphoma in heart transplant recipients is similarly difficult. The diagnosis is frequently established on post-mortem examination.103 It has also been made by endomyocardial biopsy in which the EBV genome was demonstrated in the lymphoid infiltrates.104 These infiltrates consisted of a mixture of small lymphocytes, plasma cells, immunoblasts and atypical immunoblast-like cells. They are different from the infiltrates of cellular rejection seen on previous biopsies.
Prevention
Prevention depends on limiting patient exposure to excessive immunosuppression. Lower doses and the rapid tapering of tacrolimus may limit the development of PTLD. In a review of 82 children who received renal allografts with tacrolimus-based regimens, the incidence of PTLD fell from 17% to 4%.105 This was attributed in part to a policy of aggressive tapering of tacrolimus and corticosteroids with chronic target trough concentration of 5–9 ng/mL.
Suppression of primary EBV infection or detection and early treatment may minimise the development of PTLD, especially in children.106–108
These approaches were evaluated in 40 children receiving a liver allograft.106 Among 18 high-risk children, there were no cases of PTLD and one case of EBV infection (which resolved). Among 22 low-risk patients, two cases of PTLD occurred, both of which resolved after tacrolimus was stopped; there was one case of EBV increase, which also resolved, and a 50% reduction in the incidence of PTLD.
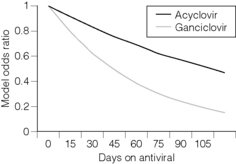
 A retrospective multicentre case–control study of adult and paediatric renal transplant recipients found that prophylactic antiviral therapy with acyclovir or ganciclovir reduced the risk of PTLD.109 The reduction was greatest with ganciclovir. Of 100 biopsy-confirmed cases and 375 matched controls, the risk of PTLD during the first year post-transplant decreased by 38% for every 30 days of treatment with ganciclovir (Fig. 12.2).
A retrospective multicentre case–control study of adult and paediatric renal transplant recipients found that prophylactic antiviral therapy with acyclovir or ganciclovir reduced the risk of PTLD.109 The reduction was greatest with ganciclovir. Of 100 biopsy-confirmed cases and 375 matched controls, the risk of PTLD during the first year post-transplant decreased by 38% for every 30 days of treatment with ganciclovir (Fig. 12.2).
A randomised trial showed that the addition of immune globulin to ganciclovir did not provide additional benefit to ganciclovir alone among patients at increased risk for PTLD.110 In one large retrospective database study, the use of prophylaxis with anti-CMV immunoglobulin during the first 4 months after kidney transplantation significantly reduced the incidence of PTLD during the first year post-transplant, but not in the subsequent 5 years.111
Treatment
Reduction in immunosuppression
Many polyclonal lymphoproliferative disease lesions resolve or improve significantly with reductions in immunosuppression.112–116
The response is best among patients with early-onset disease in whom immunosuppression is a major risk factor; in comparison, patients with late-onset or extensive disease are much less likely to benefit.82,115,117 The optimal immunosuppression reduction regimen to ensure regression of disease is unknown. The regimen adopted is based upon the severity of the disease in combination with the health risk associated with the possible loss of the allograft.
Among the severely ill with extensive disease, one regimen is reduction of prednisone to a maintenance dose of 7.5–10 mg/day and stopping all other immunosuppressive agents.102 If there is no response (as defined by a decrease in tumour mass by 10–20 days), additional therapeutic options can be entertained. Among those less severely ill with only limited disease, one regimen is the reduction by at least 50% of calcineurin inhibitor and prednisone, and the discontinuation of the antimetabolite. After 2 weeks, another 50% reduction of immunosuppression can be considered if necessary.
One report evaluated 42 transplant recipients (which included kidney, heart, lung and liver allografts) with PTLD. All were treated with a reduction in immunosuppression and 12 also underwent surgical removal of all apparent disease.115 Overall, 31 (74%) achieved complete remission. Among those treated with reduced immunosuppression alone, 63% had a complete or partial response with a median time to documentation of response of 3.6 weeks. Twelve developed acute rejection, but only one lost the graft because of rejection.115 At a median follow-up of almost 3 years, 55% of patients were alive and 50% were in complete remission.
Antiviral therapy
Many early treatment algorithms for PTLD included antiviral therapy in an attempt to control EBV infection. Acyclovir inhibits viral DNA polymerase, and in clinical studies has been shown to decrease oropharyngeal shedding of EBV.118 However, latent or transformed forms of EBV are not susceptible to antiviral agents in vitro119 and there are no in vivo data to support therapeutic acyclovir for EBV-associated PTLD.120 There is as yet no evidence of the efficacy of antiviral therapy and no consensus exists as to whether acyclovir or ganciclovir is preferred.102 Thus, although immunological and antiviral therapy have been moderately effective for treating EBV-associated infections in the lytic phase, they have been less useful in the more common latent phase of the disease and for PTLD. The lack of viral thymidine kinase expression in EBV-positive tumour cells, due to viral latency, makes antiviral therapy alone ineffective as an antineoplastic therapy. However, given the low toxicity, most centres include either acyclovir or ganciclovir with decreased immunosuppression and other therapies in the treatment of PTLD.121
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


