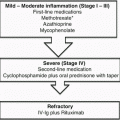
Fig. 8.1
Outline of treatments for EBA
Colchicine
Colchicine therapy is used to treat gout and has a relatively benign side effect profile. It has been used as an initial treatment for EBA with reasonable success [45]. Colchicine is a microtubule inhibitor that has the additional therapeutic benefit of down-regulating autoimmunity and inhibiting antigen presentation to T-cells [46]. Colchicine causes dose-dependent diarrhea. Therefore, it is started at 0.6 mg/day and increased as tolerated. In practice, we give 0.6 mg per day for 1 week and then increase it to 0.6 mg twice a day for a week. If there are no gastrointestinal problems, we then increase it to 0.6 mg three times a day for another week. The dosage of the prednisone is increased in this fashion each week until the patient experiences diarrhea, and then subsequently decreased by one colchicines tablet per day to the previously tolerated maximum dosage for the patient. In general, for colchicine to be effective, a dose greater than 1.8 mgs per day must be reached. About a quarter of patients with EBA have associated inflammatory bowel disease (IBD). Given the gastrointestinal side effects of colchicine, we do not use colchicine in EBA patients who have associated IBD.
Prednisone and Non-steroidal Immunosuppressive Agents
Other autoimmune bullous diseases such as BP and pemphigus are often well controlled with immunosuppressive agents such as systemic corticosteroids in combination with other potent immunosuppressive agents (methotrexate, azathioprine, cyclophosphamide, cyclosporine A, and mycophenolate mofetil). Unfortunately, EBA’s response to these measures in less predictable than other autoimmune immunobullous diseases, particularly with the non-inflammatory classical mechanobullous presentation of EBA. If these agents are tried, prednisone 1–2 mg/kg is given once a day after the patient has had breakfast alone or in conjunction with non-steroidal immunosuppressive agents. Prednisone may be used in conjunction with oral methotrexate (10–50 mg per week), oral mycophenolate mofetil (1000–3000 mg daily), oral azathioprine (50–250 mg per day based on TPMT levels), or oral cyclophosphamide (50–250 mg per day). Mycophenolate mofetil is our first non-steroidal immunosuppressive agent of choice because it appears, relative to other immunosuppressive agents, to have a lower incidence of side effects. These measures may be useful to some degree at controlling the inflammatory BP-like presentation of EBA.
Dapsone
Dapsone is an antibiotic that has the secondary property of inhibiting the migration of inflammatory cells in the skin, particularly neutrophils. A small subset of EBA patients has a neutrophil rich inflammatory infiltrate and some EBA patients have responded positively to treatment with dapsone [50]. Dapsone is given orally starting with 50 mg per day and going up as high as 300 mg per day if needed to control the disease. All patients on dapsone will get a methemoglobinemia and a concomitant drop of 1–2 g in their hemoglobin is not uncommon. For that reason, the doses of dapsone are increased slowly such that the patient can adjust and tolerate the iatrogenic anemia. Prior to starting dapsone, the patient should be evaluated for a deficiency in glucose-6-phosphate dehydrogenase (G6PD), which is a genetic disorder in which the patient has a predisposition toward hemolytic anemia. Dapsone is contraindicated in G6PD deficient patients. A simple blood test for plasma G6PD levels should be done on all patients prior to starting dapsone. Rare side effects of dapsone include bone marrow suppression, a chemical hepatitis, and drug reaction with eosinophilia and systemic symptoms (DRESS). Although these side effects may occur anytime during the patient’s course on dapsone, usually they occur early in treatment. For that reason, it is important to obtain frequent complete blood counts and comprehensive metabolic panels on patients who are on dapsone.
Rituximab
Although the number of cases of EBA treated so far is small, it appears that rituximab, a monoclonal anti-CD20 antibody, can control some patients with recalcitrant EBA refractory to other therapies [51–56]. In these reports, rituximab was often given in conjunction with other immunosuppressive agents, which were then tapered when the patient’s EBA came under control. McKinley and co-workers [49] treated a pediatric patient with EBA and achieved a sustained clinical response even after rituximab was discontinued. In general, it appears that EBA has a better prognosis in children than in adults. Rituximab is given intravenously at a dose of 375 mg/m2 of the patient’s body surface at weekly intervals for a total of 4 weeks, the same regimen as that given for a B cell lymphoma. For connective tissue diseases, rheumatologists administer rituximab intravenously at a dose of 1000 mg given 1 week and repeated 2 weeks later. This regimen has also been used for autoimmune bullous diseases with success, but so far it has not been used in EBA patients. Although rituximab may be beneficial for controlling EBA, one problem with the reported cases to date is that the EBA patients had been given prior immunosuppressive agents and were usually continued on immunosuppressive agents in addition to rituximab when control of their EBA occurred. It is not clear if monotherapy with rituximab would be beneficial for EBA.
Photopheresis and Plasmapheresis
Extracorporeal photochemotherapy (ECP), or photopheresis, has reportedly been successful in treating a number of autoimmune bullous conditions. Gordon and colleagues (58) studied three patients with recalcitrant EBA who were treated with ECP. The patients were given 1.5 mg/kg of oral crystalline 8-methoxypsoralen 90 min prior to the photopheresis treatments with a Therakos UVAR machine. Treatments were given on two consecutive days every month for a total of 6 or 7 months and then followed for 6 months post treatment. In these patients, ECP led to an improvement in clinical symptoms, an increase in dermal-epidermal adherence as measured by suction blister times, and a decrease in the level of circulating anti-BMZ antibodies [57]. There is another case report of a patient with life-threatening EBA who responded favorably and was put into remission with ECP [58]. In another report, plasmapheresis alone resulted in lower circulating anti-C7 antibodies in the blood of an EBA patient and concomitant remission of the disease [59].
Intravenous Immunoglobulin
Intravenous immunoglobulin (IVIG) has also shown efficacy in treating EBA patients [60–67]. The usual total dose of IVIG was 2 g/kg of body weight administered intravenously in divided doses over 3–5 days each month for 9–24 months. In many of these reports, the IVIG was concomitantly administered with other systemic immunosuppressives. Often conventional immunosuppression was given first and with lack of a satisfactory response, the IVIG was added and appeared to make a significant difference in controlling the patient [67]. No serious side effects have been reported in any of the EBA patients treated with IVIG.
Conclusion
EBA is an autoimmune blistering skin disease due to auto-immunity to C7, the collagen in anchoring fibrils. These autoantibodies perturb the function of anchoring fibrils leading to epidermal-dermal separation. EBA can present in a number of different ways and can be diagnosed by clinical findings, histopathology, DIF, IIF, IEM, ELISA, and Western blotting. EBA is associated with IBD and has significant clinical complications such as exuberant scarring and nail loss. EBA is notoriously difficult to treat, and there is a lack of large controlled studies, so most of the data are anecdotal case reports and small case series.
References
1.
Elliott G. Two cases of epidermolysis bullosa. J Cutan Genitourin Dis. 1895;13:10–8.
2.
3.
4.
Kushniruk W. The immunopathology of epidermolysis bullosa acquisita. Can Med Assoc J. 1973;108(9):1143–6.PubMedCentralPubMed
5.
6.
7.
8.
Woodley DT, Burgeson RE, Lunstrum G, Bruckner-Tuderman L, Reese MJ, Briggaman RA. Epidermolysis bullosa acquisita antigen is the globular carboxyl terminus of type VII procollagen. J Clin Invest. 1988;81(3):683–7.PubMedCentralCrossRefPubMed
9.
Woodley DT, O’Keefe EJ, McDonald JA, Reese MJ, Briggaman RA, Gammon WR. Specific affinity between fibronectin and the epidermolysis bullosa acquisita antigen. J Clin Invest. 1987;79(6):1826–30.PubMedCentralCrossRefPubMed
10.
11.
12.
13.
Gammon WR, Heise ER, Burke WA, Fine JD, Woodley DT, Briggaman RA. Increased frequency of HLA-DR2 in patients with autoantibodies to epidermolysis bullosa acquisita antigen: evidence that the expression of autoimmunity to type VII collagen is HLA class II allele associated. J Invest Dermatol. 1988;91(3):228–32.CrossRefPubMed
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


