, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
17.1 Vitiligo
17.1.1 Clinical Features
The worldwide prevalence of vitiligo is between 0.5 and 2 % with a high prevalence rate reported in Gujarat, India at approximately 9% [1]. Disease prevalence in the United States and Europe is estimated at 1 % with approximately half of cases beginning in childhood [1]. The incidence appears to be equal between males and females and affects all skin types. Twenty percent of individuals with vitiligo have a positive family history of the disease [2].
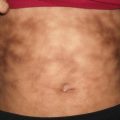
Vitiligo is characterized by white, depigmented macules and patches often occurring along sites of pressure or trauma. Mottled pigmentation and hypopigmentation can be seen at the periphery of affected areas along with islands of folliculocentric and normal coloring, suggesting repigmentation (Fig. 17.1). Depigmentation is highlighted by a Wood’s lamp fluorescence. Vitiliginous patches are usually asymptomatic, but pruritus has been reported in some cases [2]. Erythema is not frequently seen.Vitiligo can be subcategorized as localized, segmental, generalized, and universal. Segmental vitiligo, which is a variant of localized disease, often begins in childhood, most commonly along a trigeminal dermatome with associated poliosis [2]. Skin changes are typically symmetric and progressive. Universal vitiligo is a variant that affects > 80 % of body surface area, and portends the worst prognosis [2]. Segmental vitiligo, on the other hand, is stable over time and usually responds well to therapeutic interventions .
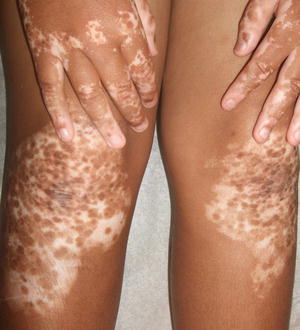
Fig. 17.1
Widespread depigmented macules and patches with islands of folliculocentric normopigmentation are seen in a patient with vitiligo
17.1.2 Histology
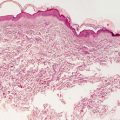
Vitiligo in children demonstrates similar histologic changes to those described in adults [3]. The histologic changes are quite subtle. The epidermis appears essentially unremarkable, although on closer examination, a loss of basilar melanocytes may be apparent (Figs. 17.2 and 17.3). The dermis is characterized by a superficial perivascular infiltrate of lymphocytes with scattered cells extending into the lower portions of the epidermis. Pigment incontinence into the dermis is either absent or minimal. Older lesions lack the dermal inflammatory infiltrate. In some cases, pigment loss may be peri-follicular in distribution, which is a pattern that is reflected with regional loss of melanocytes on histologic examination [4].
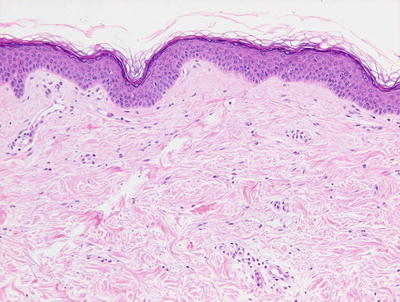
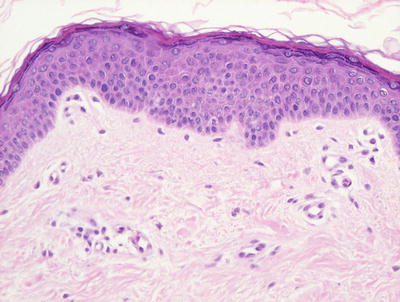
Fig. 17.2
The epidermis is largely devoid of melanocytes and melanin pigment in the basal layer in vitiligo
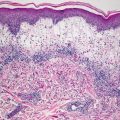
Fig. 17.3
There is lack of melanocytes along long stretches of involved epidermis in vitiligo . There is no pigment incontinence within the dermis
The histologic differential diagnosis of vitiligo includes normal skin. In many cases, it is difficult if not impossible to detect the loss of basilar melanocytes on routine H&E stains. In these situations, S100 or Melan-A stain for melanocytes may be helpful in detecting the loss of melanocytes, especially if adjacent uninvolved skin is present within the biopsy for comparison (Fig. 17.4). Other interface dermatoses can also enter the differential diagnosis, but they are readily distinguished based upon the clinical history and other characteristic histologic changes that are not seen in vitiligo.
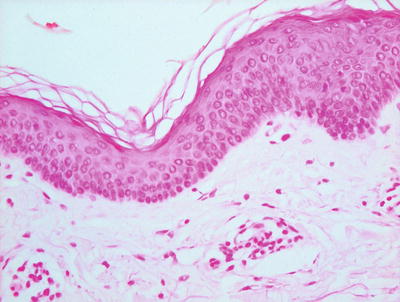
Fig. 17.4
Melan-A stain highlights the total loss of melanocytes in lesional vitiligo skin
Another differential diagnosis is post-inflammatory hypopigmentation or depigmentation. Fontana-Masson stain for melanin pigment is helpful in making this distinction (Fig. 17.5). In vitiligo, there is significant reduction in melanin pigment, whereas in post-inflammatory pigmentary alterations, melanin is present in macrophages or free in the papillary dermis. Nevus depigmentosus is also a differential consideration, but it has residual melanocytes, albeit diminished in number, while in established vitiligo, melanocytes are not present in lesional skin .
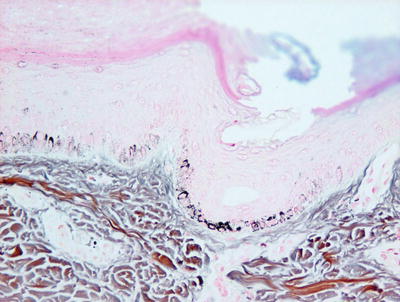
Fig. 17.5
Fontana-Masson stain demonstrates areas of reduction in epidermal melanin pigment in vitiligo
17.1.3 Pathogenesis
Vitiligo is an acquired pigmentary disorder. Most authorities now support a “convergence theory ,” highlighting a multifactorial etiology with chronic autoimmunity serving as a source of melanocyte destruction and subsequent functional impairment [2]. Research on vitiligo has been focused on understanding the initial triggering mechanism , immune propagation, and cytotoxicity against dermal melanocytes. Multiple genetic loci have been linked with vitiligo susceptibility. Many of the genes present near or in these loci encode immunoregulatory or melanocyte proteins that likely serve as autoantigens for melanocyte-specific immune response [5]. Genome-wide association studies (GWAS) identified multiple genetic risk factors for vitiligo, including genes in innate and adaptive immunity. Immune response genes implicated in vitiligo include IFIH1 and NLRP1 (genes that activate innate immune responses) and TICAM1 (an adaptor protein in toll-like receptor signaling) [6, 7]. Adaptive immune response genes implicated in vitiligo include class I and class II HLA molecules (T cell antigen recognition), PTPN22 (T cell signaling), CD80 (T cell activation), IL2Ra (T cell activation/regulation), GZMB (T cell cytotoxicity), FoxP3, and BACH2 (Treg development and function) [8]. Together, these genes constitute an immunoregulatory network that may mediate vitiligo susceptibility.
Numerous studies have implicated the role of antigen-specific CD8+ T lymphocytes in the immune-mediated destruction of melanocytes in vitiligo. Lesional skin from patients with vitiligo contains an infiltration of CD8+ T lymphocytes present adjacent to dying melanocytes in the epidermis [9]. Interestingly, the frequency of CD8+ T lymphocytes that are capable of destroying melanocytes, and the number of these cells in the blood of vitiligo patients correlate with disease severity [10, 11]. CD8+ T lymphocytes from vitiligo patients are capable of inducing melanocyte apoptosis, indicating the important role of CD8+ T cells in the pathogenesis of vitiligo [12]. Several candidate antigenic proteins in vitiligo include gp100, Mart-1, tyrosinase, and tyrosinase-related proteins 1 and 2 [13–15].
Cellular stress in melanocytes is important in disease pathogenesis. Melanocytes from vitiligo patients are intrinsically abnormal since they are more sensitive to exogenous stressors [16, 17]. They are susceptible to oxidative stress as evident by dilated endoplasmic reticulum and elevated levels of oxidative byproducts [18]. Exogenous stressors, such as monobenzone and other phenols commonly found in commercial products (e.g., rubber, leather, and cosmetic dyes), are known to both induce and exacerbate vitiligo [19, 20]. Reactive oxygen species (ROS) and heat-shock proteins generated by melanocytes in response to stress can produce damage-associated molecular patterns (DAMPs) that activate innate immune response to induce inflammation. Transcriptome analysis of skin from vitiligo patients reveals an innate immune signature particularly that of natural killer (NK) cells, and lesional skin in these patients has more NK cells than skin from healthy individuals [21].
In light of the key role of the immune system in melanocyte destruction in this disorder, combined therapeutic approaches that target both immunosuppressive and immunoregulatory responses in order to inhibit melanocyte destruction, while including regenerative approaches to promote melanocyte repopulation of depigmented regions would be an important therapeutic avenue to explore [5, 7].
17.2 Piebaldism
17.2.1 Clinical Features
Poliosis at the frontal scalp (“white forelock”) is a hallmark of piebaldism and is seen in 80–90 % of reported cases. Additionally, hyperpigmented macules at the periphery of affected areas as well as central islands of hyperpigmentation may be seen, thus distinguishing it from other achromic skin conditions [22, 23]. The disorder has a benign course, although treatment of leukoderma may be difficult.
17.2.2 Histology
Piebaldism is a clinical diagnosis, and does not ordinarily require biopsy for diagnosis. The histologic findings are subtle, and include changes in melanocytes that are not readily detected on routine histologic sections. Electron microscopy demonstrates areas of nonfunctional melanocytes, and areas of melanocytes that synthesize abnormal melanosomes [24]. In depigmented areas, epidermal melanocytes are either absent or only minimally present. There are increased numbers of both Langerhans cells and mast cells within the epidermis [25].
17.2.3 Pathogenesis
Piebaldism is an autosomal dominant disorder, and has been linked to inactivating mutations or deletions of c-Kit gene or SLUG gene [26–28]. Loss-of-function mutations of c-Kit or SLUG result in impaired melanoblast development and decreased melanogenesis. The severity of piebaldism phenotype reflects the type of c-Kit mutation. Missense mutations in the tyrosine kinase catalytic domains of c-Kit result in impaired signal transduction activity, and cause a more severe phenotype [27]. On the other hand, missense mutations in the c-Kit ligand-binding domain result in a milder form of piebaldism. SLUG is a zinc-finger transcriptional factor [29]. It is important for melanoblast migration and survival. Some patients with piebaldism were found to have heterozygous deletions of SLUG coding region [26].
17.3 Idiopathic Guttate Hypomelanosis
17.3.1 Clinical Features
Idiopathic guttate hypomelanosis is not commonly seen in children, although cases in individuals as young as 3 years old have been reported [30]. The condition frequently occurs symmetrically on sun-exposed areas of the distal arms and legs. It is characterized by 2–5 mm “porcelain-white” hypopigmented macules [30]. Younger patients have a higher frequency of facial lesions. Skin changes tend to persist once present.
17.3.2 Histology
Biopsies from hypopigmented lesions demonstrate hyperkeratosis overlying an atrophic epidermis in a few cases. Flattened rete ridges are also seen in some cases. More commonly, basal keratinocytes from affected areas of skin demonstrate decreased melanin when compared with non-lesional basal keratinocytes in the same individual (Fig. 17.6). On electron microscopic examination, degenerating melanocytes and fewer melanocytes are observed [30].
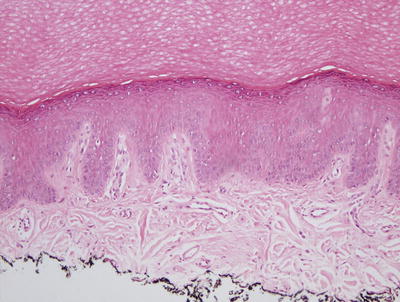
Fig. 17.6
Hyperkeratosis overlies an epidermis with marked reduction in melanocytes and little to no basilar melanin in idiopathic guttate hypomelanosis
17.3.3 Pathogenesis
The cause of idiopathic guttate hypomelanosis is not known. However, actinic influences over years of sunlight exposure may play a role in its development [31, 32]. Significantly fewer dopa oxidase-positive melanocytes are seen in idiopathic guttate hypomelanosis lesions. Reduction in the number of melanocytes and melanosomes as well as increased melanocyte degeneration has been observed. The condition may have a genetic basis. Arrunategui et al. have reported statistically significant association of HLA-DQ3 with idiopathic guttate hypomelanosis in a group of renal transplant patients [33].
17.4 Albinism
17.4.1 Clinical Features
Oculocutaneous Albinism (OCA) is an autosomal recessive disorder of impaired melanin biosynthesis. Albinism affects 1 in 17,000 people worldwide [34]. OCA Type 1 has a prevalence of 1 in 40,000, and it is seen more commonly in individuals of European ancestry [34]. Conversely, OCA Type 2 is the variant seen most commonly in those of African heritage with a prevalence of 1 in 10,000 amongst African-Americans versus 1 in 36,000 in the general population. Similarly, OCA Type 3 is seen infrequently in Caucasians and Asians, but has a prevalence of 1 in 8,500 in Africans. Oculocutaneous albinism-4 (OCA-4) is a common variant seen in Japan [26]. In all variants of OCA, patients share some degree of pigmentary dilution, congenital nystagmus, iris hypopigmentation, foveal hypoplasia, impaired visual acuity with color vision deficits, photophobia, and strabismus [34].
In OCA Type 1A, hairs at the scalp, eyebrows, and eyelashes are snow-white, and the skin does not tan. Irises are light blue and fully translucent with prominent red reflex. Ocular symptoms are more severe with poor vision and pronounced photophobia [34]. Patients may develop amelanotic nevi. In OCA Type 1B, hair and skin may develop pigmentation after infancy, and eye coloring may darken from blue to green/brown [34]. OCA Type 2 and Type 4 share the same clinical phenotype. Affected individuals have milky white to yellow-colored skin, light brown or blonde hair with varied colored irises. Patients may develop nevi and ephelides. OCA Type 3 patients have red hair and reddish brown skin. Ocular symptoms frequently are not detectable. Temperature-sensitive OCA (OCA1TS) is a form of OCA in which patients have white hair and skin at birth. At puberty, they gradually develop darker hair at cooler areas of the body, such as the extremities, but retain white hair in warmer areas, such as the scalp and axilla [26, 35]. The incidence of skin cancer is increased in individuals with albinism, thus necessitating adherence to consistent photoprotection [34].
17.4.2 Histology
Skin biopsies from patients with all forms of oculocutaneous albinism demonstrate marked decrease in melanin pigment both within the epidermis and the follicular epithelium. Melanocytes appear normal in size and shape [36]. A Fontana-Masson stain for melanin pigment highlights the absence of melanin within the biopsy specimen, but this is rarely required for diagnosis.
17.4.3 Pathogenesis
Oculocutaneous albinism is known to be associated with four genes: TYR, OCA2, TYRP1, and MATP (also called SLC45A2 ) [37]. OCA Type 1 is due to mutations in tyrosinase (TYR), which is a type I transmembrane monophenol monooxygenase [38, 39]. Tyrosinase catalyzes multiple steps in melanin synthesis, including the critical hydroxylation of tyrosine to L-DOPA, and oxidation of L-DOPA to DOPA-quinone [40]. Mutations in TYR can cause complete or partial OCA depending on the extent of loss of TYR enzymatic activity. Patients with TYR mutations have no melanin formation throughout their life because the first step of melanin synthesis is blocked.
Oculocutaneous albinism-2 (OCA2) is due to mutations in the OCA2 gene, which is an integral membrane transporter protein that regulates the processing and transport of tyrosinase to melanosomes [37, 41].
Oculocutaneous albinism-3 (OCA3) is caused by mutations in the TYR-related protein-1 (TYRP-1) gene [26]. TYRP1 is structurally similar to TYR, and it is necessary for the synthesis of black/brown eumelanin [42]. TYRP1 complexes with and stabilizes tyrosinase [43]. In the absence of TYRP1, tyrosinase is rapidly degraded.
Oculocutaneous albinism-4 (OCA4) is due to mutations of TYRP-1 and membrane-associated transporter protein (MATP) (also called SLC45A2 ) [26]. MATP is involved in the intracellular processing and trafficking of melanosomal proteins. Mutations in MATP cause mistrafficking of tyrosinase [37, 44, 45]. Lastly, patients with temperature-sensitive OCA have a missense mutation in the tyrosinase gene that produces a mutated enzyme, which is temperature-sensitive and exhibits some activity at 35 °C and no activity at temperature above 35 °C [46].
17.5 Chediak–Higashi Syndrome
17.5.1 Clinical Features
Chediak–Higashi syndrome is an autosomal recessive genetic disease characterized by partial albinism, recurrent pyogenic infections, and blood cell dyscrasia with subsequent severe immunodeficiency and lymphoproliferative syndrome [47]. Patients typically present with generalized pigmentary dilution and much lighter skin color than their parents or siblings [47]. They have hair with a silvery-gray sheen, and ocular symptoms of photophobia, nystagmus, and retinal pigmentary dilution [48].
The childhood form of Chediak–Higashi syndrome is the most severe, and is seen in 85–90 % of affected individuals [47]. These individuals experience an often fatal accelerated phase with diffuse lymphohistiocytic organ infiltration and death by the second decade of life. The mildest form is seen in 10–15 % of patients. In this variant, patients are not predisposed to infections, but they develop progressive and fatal neurologic sequelae. The rarest form is “adolescent” disease in which individuals present with developmental delay, hypopigmentation, and recurrent infection but no accelerated phase [47]. For those with hematologic disease, a cure can be achieved with bone marrow transplantation prior to the accelerated phase .
17.5.2 Histology
Biopsy is rarely indicated or performed in patients with Chediak–Higashi syndrome as the findings on histology are rarely contributory in establishing a diagnosis. Diminished basilar melanin has been identified in biopsies taken from depigmented areas of the skin (Figs. 17.7 and 17.8). The pattern of enlarged membrane-bound granules within neutrophils that characterizes the syndrome has also been reported in melanocytes and Langerhans cells [49, 50].
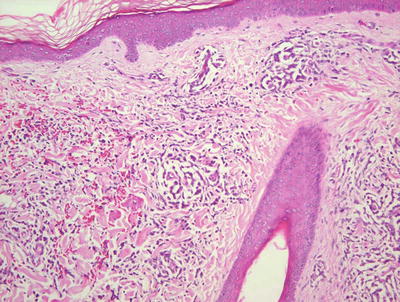
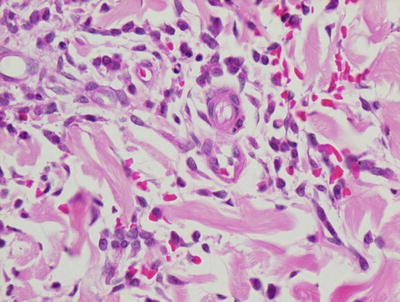
Fig. 17.7
Decreased basilar melanin is observed in the epidermis in Chediak–Higashi syndrome
Fig. 17.8
The dermis in Chediak–Higashi syndrome contains a mixed inflammatory infiltrate with abundant red blood cell extravasation
17.5.3 Pathogenesis
Chediak–Higashi syndrome is characterized by giant lysosomal and non-lysosomal inclusions in cells containing granules, such as melanocytes and hematopoietic granulocytes [26, 40]. Giant inclusions within various vesicular organelles, such as lysosomes, melanosomes, cytosolic granules, and platelet dense bodies, are observed in various cell types. The presence of giant melanosomes in melanocytes, and the abnormal distribution of these melanosomes to keratinocytes may account for the hypopigmentation phenotype in patients with Chediak–Higashi syndrome.
The causative gene is CHS1⁄LYST, which is important in intracellular vesicular transport and vesicular size [51, 52]. The mouse homolog of CHS1 is the beige gene, and studies of transgenic mice with CHS1⁄LYST overexpression provide molecular insights into the role of this molecule. Mice with overexpression of the affected gene beige have smaller lysosomes as compared with wild-type mice. A screen for CHS1-binding partners using a yeast two-hybrid system identified several interacting proteins in vesicle trafficking, including hepatocyte growth factor-regulated tyrosine kinase substrate (HRS), 14-3-3, casein kinase II, and LIP5 [53, 54]. These proteins are involved in protein sorting and trafficking to the lysosome. Loss of CHS1⁄LYST may result in abnormal incorporation of lysosomal proteins into lysosomal membranes, or ineffective removal or recycling of these proteins in membrane trafficking .
17.6 Incontinentia Pigmenti
17.6.1 Clinical Features
Incontinentia pigmenti is a multisystem disorder of ectodermal origin with extracutaneous manifestations affecting the dental, ocular, and central nervous systems. As of 2013, there were 800 registered cases of incontinentia pigmenti with global incidence of 1:40,000 [55]. Incontinentia pigmenti overwhelming affects females, and is usually fatal to males in utero.
Cutaneous manifestations classically present in four successive stages, although they may overlap or not even occur [56] (Figs. 17.9 and 17.10). The stages are (stage 1) erythema followed by blaschkoid vesicles and pustules within the first 2 weeks of life, typically clearing by 4 months of age; (stage 2) hyperkeratotic to verrucous papules and linear plaques; (stage 3) linear hyperpigmentation; and (stage 4) hypopigmentation and possible scarring [55].
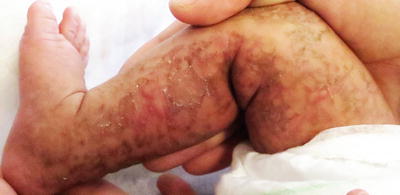
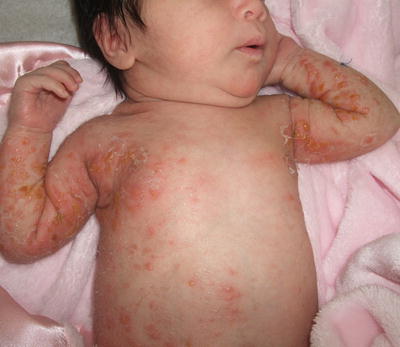
Fig. 17.9
Hyperkeratotic papules and reticulate hyperpigmentation in a Blaschkoid distribution are present on the leg of an infant with incontinentia pigmenti
Fig. 17.10
Incontinentia pigmenti in this infant presents as Blaschkoid plaques comprised of erythematous papules, vesiculopapules, and yellow-colored vesicles, mostly concentrated on the extremities and lateral torso typifying stage 1 of the disease
Additional clinical features include dental anomalies, such as partial anodontia and conical teeth, which can be seen in as many as 70 % of affected individuals [55, 56]. Patients may also present with nail dystrophy and alopecia along the vertex of the scalp. Ocular disease can be seen in 20–40 % of patients with complications including retinal disease, strabismus, cataract, and pigmentation of the conjunctiva [55]. The greatest morbidity and mortality are associated with neurologic disease, which occurs in approximately one-third of patients with infantile seizures as the most common finding. Eight percent of patients will have some degree of mental retardation.
17.6.2 Histology
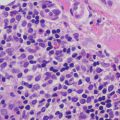
The histologic changes seen in incontinentia pigmenti are dependent upon the stage of the lesion biopsied. In the earliest stage 1 lesions (vesicular stage), the characteristic findings are those of an “eosinophilic spongiosis” (Fig. 17.11). The epidermis is markedly spongiotic, often with coalescing spongiotic microvesicles [57]. Focal necrosis within the epidermis is present, and widely scattered apoptotic keratinocytes are easily apparent both within the epidermis and follicular epithelium (Fig. 17.12). The dermis demonstrates lymphocytes and eosinophils around the vessels of the superficial vascular plexus and extending into the epidermis.
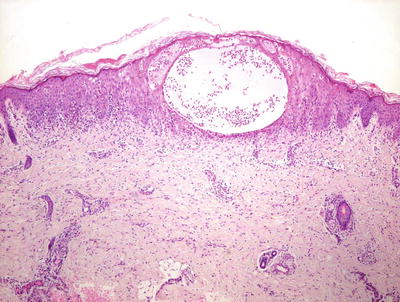
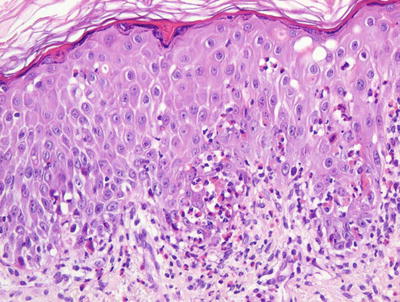
Fig. 17.11
Early stage lesions of incontinentia pigmenti show intraepidermal vesicles, abundant exocytosis, and scattered dying keratinocytes
Fig. 17.12
Higher magnification shows abundant eosinophils within the dermis and epidermis and multiple dying keratinocytes that characterize incontinentia pigmenti
Stage 2 (verrucous stage) lesions demonstrate an acanthotic epidermis, with marked overlying hyperkeratosis. Dyskeratotic cells are prominent within the epidermis, and a whorled configuration to these cells has been described. Epidermal eosinophilia is no longer a prominent feature in lesions that have progressed to the verrucous stage, but can still be identified focally.
Stage 3 lesions are essentially those of post-inflammatory pigmentary alteration. The prominent histologic changes include loss of epidermal pigment with abundant accumulation of melanin within melanophages in the papillary dermis. Focal basal vacuolization and dyskeratosis of basal keratinocytes are present. In this stage, the lesions are no longer diagnostic but require clinical correlation in order to arrive at the correct diagnosis. In the late Stage 4 lesions, there is epidermal atrophy and a variable reduction in the number of melanocytes.
Twenty-nail dystrophy has been described in patients with incontinentia pigmenti with hyperkeratosis and hypergranulosis. Pseudoepitheliomatous hyperplasia is present with abundant dyskeratotic cells, similar to the findings in the verrucous stage of cutaneous lesions [58].
17.6.3 Pathogenesis
Incontinentia pigmenti is a rare genodermatosis that segregates as an X-linked dominant disorder. Females with incontinentia pigmenti exhibit skewed X-inactivation because cells expressing the mutated X chromosome are eliminated selectively in utero. Although the majority of patients are females, male patients have been identified. The survival of males is linked to hypomorphic NEMO mutations that have residual NEMO and NF-κB activities and therefore are not lethal [59, 60]. Males with incontinentia pigmenti tend to have additional medical features not usually associated with the disease, including immune dysfunction, osteopetrosis, and lymphedema.
A landmark study showed that the cause of incontinentia pigmenti is due to genomic rearrangement in NEMO (nuclear factor-κB essential modulator, also known as inhibitor κB kinase, γ-subunit) [61]. Mutation in NEMO, resulting in the deletion of exons 4 through 10, has been found to account for more than 80 % of all incontinentia pigmenti cases. NEMO is the regulatory subunit of the inhibitor κB kinase (IKK) complex, and it is essential for the activation of the transcription factor NF-κB [62, 63]. Loss of NEMO disrupts NF-κB activity in the cell. As a consequence, incontinentia pigmenti cells are highly sensitive to pro-apoptotic signals. Genetically engineered mice with loss of NEMO develop a phenotype closely resembling that of incontinentia pigmenti in humans, including generalized erythematous vesicular eruptions, followed by hyperkeratotic scaly lesions and lines of altered pigmentation. Histologic examination of NEMO-knockout mice showed eosinophil- and neutrophil-rich intraepidermal microvesicles with spongiosis and dyskeratotic keratinocytes [64, 65]. Conditional deletion of NEMO only in skin keratinocytes shows that loss of NEMO in these cells is sufficient to induce inflammatory skin lesions, indicating that expression of NEMO in keratinocytes is a potent initiator of skin inflammation in incontinentia pigmenti [66].
Another key player in the pathogenesis is eotaxin, which is an eosinophil-selective chemokine that has been isolated from the blister fluid and crusted scales of patients with incontinentia pigmenti [67, 68]. Eotaxin has a NF-κB binding site. Lesional skin from patients shows strong expression of eotaxin in the epidermis by immunohistochemical staining, and eotaxin expression corresponds to areas of eosinophil accumulation in the epidermis [69]. The peripheral eosinophilia observed in incontinentia pigmenti is due to increased production of eosinophils in the bone marrow, and increased migration of eosinophils into the circulation. Granulocyte-monocyte colony-stimulating factor (GM-CSF) induces eosinophil differentiation and maturation in the bone marrow, and is also released by activated eosinophils, thus providing a feedback regulatory loop for eosinophil activation [69].
17.7 Xeroderma Pigmentosum
17.7.1 Clinical Features
Xeroderma pigmentosum is an autosomal recessive disorder with 8 complementation groups and a variant subtype [70]. There is an estimated disease prevalence of 1 per 1,000,000 individuals in the United States and Europe [71]. Prevalence is greater in North Africa and the Middle East in populations with higher incidence of consanguinity. Additionally, the number of cases is higher in Japan with reported prevalence of 1 in 22,000 individuals [71].
Xeroderma pigmentosum is characterized by severe photosensitivity, ocular disease, and increased risk of skin malignancy. Approximately 50 % of individuals have an acute sunburn reaction with erythema and blistering following minimal UV light exposure [72]. They may have pronounced photodistributed freckling before 2 years of age (Fig. 17.13). With continued sun exposure, patients develop poikiloderma, actinic keratosis, and dry parchment-like hyperpigmentation of the skin [73]. Patients are predisposed to premature accelerated photoaging with 1000-fold increased risk of skin malignancies with many patients developing their first skin cancer by 10 years of age.
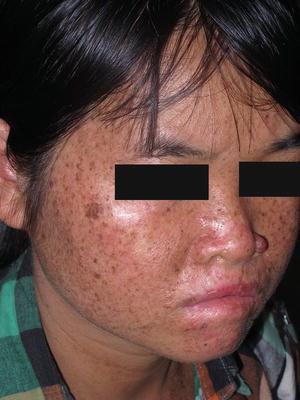
Fig. 17.13
Xeroderma pigmentosum presents with pronounced photodistributed freckling and poikiloderma. Scleral injection and actinic cheilitis also can occur as seen in this patient (photo courtesy of Minh Hoang, MD, Ho Chi Minh City, Vietnam)
17.7.2 Histology
The diagnosis of xeroderma pigmentosum cannot be established based upon skin biopsy alone. The earliest observable skin changes include epidermal atrophy and telangiectasia. These findings are not specific, but they are unusual findings in children and suggest abnormal skin. Clumping of melanin pigment in the basal layer is another early but nonspecific finding [74]. Papillary dermal fibrosis representing early scarring is also a preliminary finding.
Patients with xeroderma pigmentosum are at a greatly enhanced risk for developing a myriad of cutaneous neoplasms. The most frequent include squamous cell carcinoma and basal cell carcinoma [75–77]. Basosquamous carcinomas have also been described [78]. The risk for melanoma is also greatly increased [79]. Atypical fibroxanthoma, which is a tumor ordinarily associated with elderly individuals, arises in patients with xeroderma pigmentosum [77, 80]. Cutaneous appendageal tumors also appear with increased frequency in this population [81, 82]. The histologic findings in each of the aforementioned malignancies are identical to those seen in individuals without underlying xeroderma pigmentosum. The difference is that in this population, these tumors occur at a very early age, while in the general population, they are tumors associated with sun-damaged skin occurring in an elderly population .
17.7.3 Pathogenesis
The pathogenesis of xeroderma pigmentosum is due to defective mechanism to repair UV radiation-induced damage to DNA [83, 84]. In normal cells, UV-induced DNA damage is repaired by the nucleotide excision repair (NER) pathway and double-strand breaks are repaired by homologous recombination or nonhomologous end-joining [85, 86]. Due to defects in DNA repair, key genes that regulate genomic stability (such as the p53 tumor suppressor gene) are mutated earlier and more frequently in these patients.
There are at least eight complementation groups (genetic subtypes) of xeroderma pigmentosum (XP-A to XP-G, plus XP variant) with each subtype associated with at least a unique gene defect [87, 88]. These groups are associated with defects in one of the many proteins involved in NER process, which is the primary mechanism by which UV-induced adducts in cellular DNA are removed. The genes involved in some of these groups have been identified. The helicases ERCC3 (encoded by XPB gene), ERCC2 (encoded by XPD gene), and ERCC5 (encoded by XPA and XPG genes) are involved in the unwinding of DNA in the region of damaged DNA [87]. DDB2, which is the XPE gene product, recognizes UV-induced DNA damage and marks it for repair. The endonucleases ERCC4 (encoded by XPF) and ERCC5 (encoded by XPG) incise DNA strand and excise small fragments of the damaged genome for repair [89, 90]. Mutations in any of the proteins involved in the “transcription coupled DNA repair (TCR) ” pathway, the global genomic repair (GGR) pathway, or the common nuclear excision repair pathway lead to genomic instability and cancer predisposition as observed in xeroderma pigmentosum. There are some cases called XP variant (XP-V) group, which are characterized by a defect in “trans-lesion DNA synthesis.” With these mutations, the DNA polymerase-eta that synthesizes DNA past these adducts is defective, allowing synthesis by DNA polymerases that are more “error prone,” thus introducing errors during DNA replication [91, 92].
17.8 Poikiloderma Congenitale (Rothmund Thomson Syndrome)
17.8.1 Clinical Features
Poikiloderma congenitale (Rothmund-Thomson syndrome) is a rare autosomal recessive disorder [93]. Rothmund–Thompson syndrome is characterized by poikiloderma, sparse hair at the scalp, eyebrows, and eyelashes, small stature, skeletal abnormalities, cataracts, and an increased risk for cancer, specifically osteosarcoma [93]. There are two subtypes of disease, types I and II. Individuals with type I disease develop juvenile cataracts with no incidence of osteosarcoma or bone disease, while those with type II disease have skeletal abnormalities and increased risk of osteosarcoma without developing cataracts. At 3 to 6 months of age, patients often present with erythema, swelling, and blistering of the face with sparing of the trunk and intertriginous folds [93]. Skin changes evolve into poikiloderma with punctate atrophy involving the extensor extremities, trunk, abdomen, and buttocks. Hyperkeratotic plaques and edema may develop on the elbows, knees, hands, and feet in association with plantar keratoderma [73, 93, 94]. Ten to fifty percent of patients develop juvenile cataracts, usually between 4 and 7 years of age [93, 94]. Osteosarcoma is the most frequently seen malignancy with a prevalence of approximately 30 %. Cutaneous tumors are also increased in these patients with a prevalence of ~5 %.
17.8.2 Histology
Histologic changes of affected skin in Rothmund-Thomson syndrome include epidermal atrophy with marked variability in basilar melanin deposition. The rete ridges are flattened, and occasional basal vacuolization is present. Early lesions demonstrate a band-like lymphocytic infiltrate that may infiltrate the lower portions of the epidermis. In some areas, the basal layer appears to have normal amount of melanin, while melanin is markedly decreased in other areas in the epidermis. Melanophages are present within the papillary dermis and around the vessels of the superficial vascular plexus. Papillary dermal edema is seen in most cases. Vascular ectasia is present throughout the superficial portions of the dermis [95].
17.8.3 Pathogenesis
Karyotypic analysis of cells isolated from Rothmund-Thomson syndrome patients shows a high frequency of chromosomal abnormalities. RECQL4 has been identified as a causative gene [96], and it is present in 66 % of clinically diagnosed patients [97]. Rothmund–Thompson syndrome type I does not have mutations in RECQL4 gene [98], while type II patients demonstrate documented mutations in RECQL4. The identified mutations result in early termination codon in the helicase domain of RECQ4, resulting in truncation and loss of function of RECQL4 protein [96]. RECQL4 analysis showed that patients with Rothmund–Thompson syndrome who developed osteosarcoma carried at least one deleterious RECQL4 mutation, thus implicating a role for RECQL4 in osteosarcoma formation in these patients.
RECQL4 functions in several aspects of genomic stability maintenance, and plays an important role in DNA replication initiation and double-strand break repair [99, 100]. RECQL4 is a RecQ DNA helicase important in DNA replication whose function is to unwind and separate the two complementary DNA strands. The RecQ family of DNA helicases unwinds DNA in the 3′ to 5′ direction in relation to the DNA strand to which the enzyme is bound. RECQL4 also forms a stable complex with components of ubiquitin ligases, which are essential for chromosome stability [96]. Animal models in which the RECQL4 gene is deleted have a high perinatal mortality, while the survivors have severe growth retardation and developmental defects similar to the abnormalities seen in Rothmund–Thompson syndrome patients, thus strongly implicating a role for RECQL4 in disease pathogenesis [101]. Mutations in RECQL4 give rise not only to Rothmund–Thompson syndrome but also to two other known autosomal recessive diseases, RAPADILINO and Baller–Gerold syndrome. Both Rothmund–Thompson syndrome and RAPADILINO patients have increased risk for osteosarcoma and lymphoma [98].
17.9 Bloom Syndrome
17.9.1 Clinical Features
Bloom syndrome is a rare autosomal recessive disorder. Affected children are often males, and there is greater incidence in individuals of Ashkenazi Jewish ancestry [102]. Bloom syndrome is characterized by growth retardation, type II diabetes, and recurrent infections [73]. Photodistributed erythematous patches and telangiectasia along the malar region of the face, dorsum of the hands, and extensor forearms often appear within the first few years of life. Patients may also lose the lower eyelashes, and have blistering and fissuring along the lower lip. Many patients also have café-au-lait spots with hypopigmentation along the peripheral edges [73, 102]. A wide array of ocular abnormalities, narrow facies with prominent nose as well as malar and mandibular hypoplasia can also be seen [102]. Photoprotection is encouraged in these patients, although skin manifestations often improve with time. Patients are predisposed to a multitude of malignancies, including skin and hematologic cancers frequently by the second decade of life [73, 102–104] (Fig. 17.14). Males are infertile, while females are fertile but may experience early menopause [103].
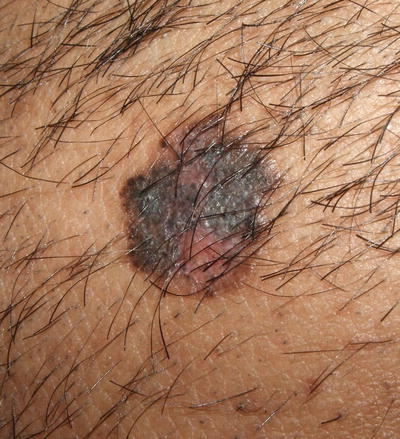
Fig. 17.14
Pigmented basal cell carcinoma in a patient with Bloom syndrome
17.9.2 Histology
Histologic features in Bloom syndrome resemble those seen in lupus erythematosus. There is interface dermatitis, and the epidermis demonstrates extensive basal vacuolization with exocytosis of lymphocytes (Figs. 17.15 and 17.16). Melanophages are present in the dermis with a moderately intense superficial perivascular lymphocytic infiltrate. Dilated capillaries are present, resulting in clinically apparent telangiectasias [105].
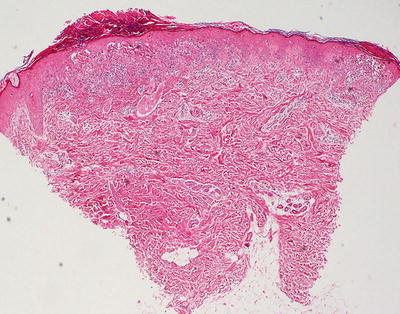
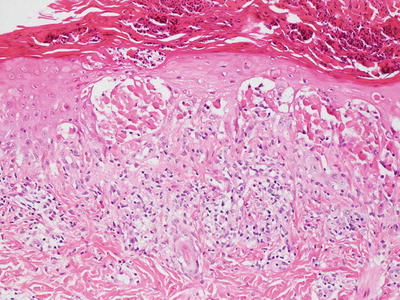
Fig. 17.15
Bloom syndrome is characterized by an interface dermatitis with abundant dying keratinocytes within the basal layer of the epidermis
Fig. 17.16
A lymphocytic infiltrate is present in the papillary dermis and extends into the epidermis with dead keratinocytes in Bloom syndrome
The histologic differential diagnosis includes lupus erythematosus and other interface processes. Unlike lupus erythematosus, basement membrane thickening and mucin deposition are not seen in Bloom syndrome skin biopsies. Direct immunofluorescence stains do not reveal immunoglobulin deposits at the dermal-epidermal junction [105]. A phototoxic drug eruption could also be considered. Clinical correlation is the best way to make this distinction because the histologic findings in these entities are identical .
17.9.3 Pathogenesis
Bloom syndrome is an autosomal recessive disease caused by mutations in the BLM gene (Bloom syndrome, RecQ Helicase-Like) [107]. BLM encodes for RecQL3 helicase, which has important function in the fidelity of DNA replication. RecQL3 is an enzyme that repairs breaks in double-stranded DNA [108, 109]. BLM also functions to suppress crossover formation between sister chromatids during DNA replication. More than 60 mutations in BLM gene have been associated with Bloom syndrome. The most common mutation is the deletion of six nucleotides at position 2281, and the insertion with seven other nucleotides at this position [110, 111]. Mutation in BLM causes a tenfold increase in the frequency of DNA exchange between sister chromatids, and prevents the removal of Holliday junctions during DNA replication, resulting in segmented and entangled sister chromatids [112]. Bloom syndrome cells have increased chromosome breaks, spontaneous mutation rates and frequency of spontaneous sister chromatid exchanges, thus contributing to genomic instability and cancer predisposition [113–115]. Patients with Bloom syndrome are 150–300 times more likely to develop malignancy than the general population [110].
17.10 Nevus Depigmentosus
17.10.1 Clinical Features
Nevus depigmentosus is present at birth or appears soon after, most commonly within the first year of life [116]. Nevus depigmentosus is characterized by an irregularly shaped, hypopigmented patch or macule that does not fluoresce with Wood’s Lamp examination. The distribution and location of a nevus depigmentosus are stable with growth proportional to that of the child. The lesion persists over a lifetime [116].
17.10.2 Histology
Histologic sections of nevus depigmentosus resemble that of normal skin on first examination. The changes are quite subtle and may only be evident with use of Fontana-Masson stain that demonstrates marked reduction in basilar keratinocytic melanin [117]. A decrease in melanocytes has been reported by some authors [117], but other authors find normal numbers of melanocytes as identified with immunomarkers [116, 118].
The differential diagnosis includes post-inflammatory hypopigmentation, but dermal melanophages are not present in nevus depigmentosus. Vitiligo can be difficult to distinguish from nevus depigmentosus, but the presence of lymphocytes with exocytosis into the epidermis favors the diagnosis of vitiligo. Low magnification suggests normal skin, and close attention to the clinical history should raise concerns for a diagnosis of nevus depigmentosus. The diagnosis is easily confirmed with Fontana-Masson stain for melanin .
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


