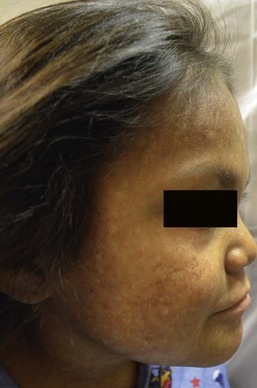
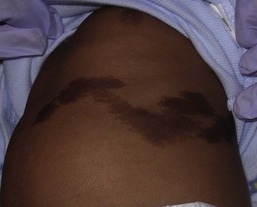
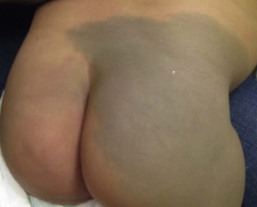
Harper N. Price, Ashfaq A. Marghoob Hyperpigmented lesions, presenting at birth and during the first few weeks of life, are quite common. Pigmented lesions can range from small and isolated to large and multiple. They can exist independently, or in association with other signs and symptoms, and may lead to the diagnosis of a congenital or genetic skin disease. In some cases, hyperpigmented ‘birthmarks’ may not be evident at birth but appear weeks to months later. The relatively light color of infant skin can make some pigmentary disorders difficult to appreciate and thus observation over time may be helpful. Also, over time, melanocytes may produce more pigment and differences between normal and hyperpigmented or hypopigmented skin may become more evident. This chapter serves as an aid for clinicians in diagnosing pigmented lesions in the neonatal and infantile period with an approach based on coloration and localization of lesions with a review of diagnostic criteria, associated conditions, and suggested clinical evaluation when applicable (Table 24.1). Café-au-lait macules (CALM) are well-circumscribed, oval to round, hyperpigmented hairless macules or patches that may be noted at birth, during infancy or throughout childhood. CALM may have the color of ‘coffee with milk’ in lighter skinned individuals but in darker skinned persons the color may be more medium to dark brown. The size of CALM ranges from a few millimeters to over 20 cm and can occur anywhere on the body (Fig. 24.1). Morphologically, the borders of CALM have been described as smooth (resembling the ‘coast of California’) to jagged contours (resembling the ‘coast of Maine’), correlating with specific disorders. However, in general, this feature varies widely and it not a reliable diagnostic sign. The prevalence of CALM varies in the general population with 2.7% of 4641 newborns having one or more macules in a US study.1 There are great differences in prevalence amongst various racial groups, with an increased number of CALM seen in darker skin types.1–4 A higher prevalence (24.2–36%) of CALM has been reported in older children and young adults as compared with infants.2,3,5 The presence of up to three CALM has been reported in 10–28% of the general population.3,6,7 Multiple CALM, especially six or more, should prompt the clinician to consider underlying disorders (Table 24.2). TABLE 24.2 Disorders associated with multiple café-au-lait macules (CALM) and associated features a Arnsmeier, Riccardi and Paller20 proposed that absence of two consecutive generations of neurofibromas and other non-pigmentary changes of NF-1 allows the diagnosis of familial CALM. b Syndromes listed have been described with CALM but these are not considered part of the diagnostic criteria, and may not be seen in all cases. Although the diagnosis is generally made through clinical examination, histologic examination of CALM shows increased melanin content in keratinocytes and melanocytes without evidence of melanocyte proliferation. On electron microscopy, giant melanosomes may be seen. While establishing the diagnosis of CALM, these microscopic features are not useful in determining specific systemic associations (such as neurofibromatosis). The differential diagnosis of CALM includes a congenital melanocytic nevus, speckled lentiginous nevus, lentigo, Becker’s nevus and forms of pigmentary mosaicism such as nevoid hypermelanosis. Often the distinction will become evident over time. Individual lesions of urticaria pigmentosa or a solitary mastocytoma may also be mistaken for CALM and can be distinguished by the Darier’s sign (the development of urticaria following firm stroking of a mastocytoma) (see Chapter 28). Treatment of isolated CALM is generally not necessary. Laser therapy, typically with ‘Q-switched’ lasers (Q-switched ruby, Q-switched alexandrite, and Q-switched Nd : YAG), has been attempted for cases considered extensive or disfiguring, but results are inconsistent and repigmentation may occur.8–11 CALM may also paradoxically darken with laser treatment and result in postinflammatory hyperpigmentation (usually transient).8 Segmental pigmentation disorder (SPD or mosaic hyperpigmentation, pigmentary mosaicism) refers to a hyperpigmented or hypopigmented patch often with a sharp midline demarcation and a less distinct lateral border, commonly located on the trunk.12 Lesions are often noted in infancy and are described in a segmental block-like pattern resembling a ‘checkerboard’13 or following the lines of Blaschko. Those lesions that less commonly involve non-truncal sites (such as the extremity or neck) usually extend onto the torso, and solitary facial involvement has been described. The majority of patients with SPD have no extracutaneous abnormalities. SPD is thought to be due to somatic mutations resulting in cutaneous mosaicism. Extensive cases can be variably associated with chromosomal anomalies. The natural history of SPD has not been reported, but persistence of the hyperpigmentation is expected. A rare biopsy done on a patient with hyperpigmented SPD showed increase melanin in the basal layer, similar to CALM.12 CALM are usually the first presenting sign in NF-1, and multiple CALM can be seen in 90% of patients with NF-1 on the trunk, buttocks and legs.14 In fair-skinned individuals, CALM may be initially overlooked on cutaneous examination. The number of CALM increases over time during the first decade of life and lesions tend to grow in proportion to the child and may darken with sun exposure. Axillary and/or inguinal freckling is usually absent at birth but present by age 3–5 years, although it may be noted earlier.15 This common feature tends to be the cutaneous sign, along with multiple CALM, that establishes the diagnosis and is felt to be pathognomonic for NF-1.16 Multiple CALM can be seen in other variants of NF, including neurofibromatosis type 2, segmental NF-1 and hereditary spinal neurofibromatosis. A more extensive discussion of NF can be found in Chapter 29. Legius syndrome (neurofibromatosis type 1-like syndrome) is a recently identified autosomal dominant disorder caused by a mutation in the SPRED1 gene.17 The SPRED1 gene is responsible for making spred-1 protein which inhibits the Ras/MAPK pathway. Affected individuals typically have CALM and axillary freckling. However, neurofibromas, typical osseous lesions, Lisch nodules of the iris and central nervous system tumors are characteristically absent. Some patients with Legius syndrome have been described with macrocephaly, a Noonan-like facial appearance, learning disorders, developmental delay and/or attention deficit/hyperactivity disorder in childhood.17,18 Several families with Legius syndrome have been described with lipomas as well.17–19 Many individuals may meet the NIH diagnostic criteria for NF-1 but gene testing will be negative for mutations in the neurofibromin gene. It is uncertain if there is an increased risk of malignancies. However, some authors have proposed less stringent monitoring in Legius patients as compared to NF-1 patients.18 Screening for developmental delay and behavioral and learning problems is recommended as well as examination and counseling by a clinical geneticist familiar with Legius syndrome. Multiple familial CALM have been described in several families without the other stigmata of NF-1, including the absence of NF-1 gene mutation. SPRED1 had not been tested in these families.20 This diagnosis should only be made in an older child without the other stigmata of NF-1 with a clear family history of multiple CALM without other signs of NF-1.4 The cardinal features of McCune–Albright syndrome (MAS) include polyostotic fibrous dysplasia, endocrinopathies and precocious puberty as well as CALM. MAS is a rare and sporadic disorder caused by a postzygotic mutation in the gene, GNAS1.21–23 The GNAS1 gene encodes for the alpha subunit of the guanine nucleotide-binding protein, causing loss of GTPase activity and increased stimulation of the adenylate cyclase system, resulting in proliferation and autonomous hyperfunction of hormonally responsive cells.24–26 This autosomal dominant mutation is only compatible with life when it occurs in individuals as a postzygotic somatic mutation with resulting mosaicism, which in turn results in marked phenotypic variability. The CALM seen in MAS are often large, and segmental with favored distribution over the torso and buttocks. They may also follow the lines of Blaschko (Fig. 24.2).27,28 The border of the CALM in MAS is often described as ‘jagged’ and irregular. These cutaneous findings may be present at birth or within the first few years of life and have been noted in approximately 53–98% of patients with MAS.29,30 Melanotic macules of the oral mucosa have also been reported.31,32 Extracutaneous findings in MAS include polyostotic fibrous dysplasia, where bone is replaced by fibrous tissue resulting in asymmetry, bony growths and pathologic fractures; this finding is rarely seen at birth and tends to manifest over time, usually within the first decade. In addition, endocrine abnormalities that may occur include precocious puberty, hyperthyroidism, Cushing syndrome, hyperparathyroidism, hyperprolactinemia, and hypersomatotropism.25,33–35 Neonatal cholestasis, acholic stools and jaundice have been described as presenting symptoms in neonates with MAS.35 Due to the variable clinical expression and segmental nature, diagnosis of MAS may be difficult in infancy. Cutaneous findings of MAS must be differentiated from NF-1. The CALM found in NF-1 tend to be smaller, lighter in color, and more scattered in distribution. Histologic studies would not be helpful in differentiating these disorders. Radiologic studies for MAS may not reveal bony abnormalities in the neonatal period and CALM often precede the development of bony changes by 3 months to 9 years.36 When MAS is suspected, close observation for endocrine abnormalities is warranted. In those with known or suspected MAS, a referral to orthopedics and endocrinology is recommended. Prognosis for MAS is generally favorable and the development of malignancy is rare. Extensive osseous dysplasia early in life, however, portends a poorer prognosis.36 The presence of dermal melanocytes in the skin can give rise to several clinically appearing blue-gray, green or black lesions on the skin including congenital dermal melanocytosis (Mongolian spots), Nevus of Ota, Nevus of Ito and blue nevi. The blue color is due to the Tyndall effect: the scatter of light passing through a turbid medium, such as the dermis, as it strikes melanin particles. Mongolian spots are common birthmarks present in over 80% of black and Asian infants compared with only up to 10% of Caucasians infants.37,38 They present at birth, or early infancy, as blue-gray, blue-green, or blue-black macules most often noted over the sacrum and gluteal regions (Fig. 24.3) and less often on extensor extremities and the posterior shoulder. Size can range from a few millimeters to over 10 cm and single or multiple lesions may be present. The borders of Mongolian spots are ill-defined and tend to fade gradually into the surrounding skin color. The coloration will often stabilize in infancy and fade during the first decade of life. Only a small number of lesions persist (3–4%) after adolescence and it has been suggested that these are likely not distinct entities but analogous to Nevus of Ota or Ito.37,39–41 Clinically, these persistent lesions are often blue-black homogenous or speckled patches present in a segmental distribution. In the Mongolian spot, elongated, slender and slightly wavy dendritic cells containing melanin granules are found in the mid and deep dermis.42 These dendritic melanocytes are present in low numbers and scattered between collagen bundles, both of which are orientated parallel to the skin surface. There is an absence of melanophages. The reason for regression of Mongolian spots and failure of regression of other dermal melanocytic lesions such as nevus of Ito or Ota with age is currently unknown. Mongolian spots are common, and associated abnormalities are rare. Dermal melanocytosis may be associated with certain lysosomal storage diseases, including gangliosidosis type 1 (GM1), Niemann–Pick disease, Hunter syndrome, α-mannosidosis syndrome, and Hurler syndrome.43 Dermal melanocytosis is generally extensive in distribution often involving the ventral and dorsal aspects of the trunk in addition to the sacrum and extremities.44–50 Mongolian spots may also co-exist within CALM in NF-1.51,52 Dermal melanocytosis is part of the criteria for a subgroup of neurocristopathies labeled ‘phakomatosis pigmentovascularis’ (PPV), in which vascular malformations of the skin, pigmented nevi and Mongolian spots occur together in the same patient (see below). Cleft lip malformation has been associated with adjacent dermal melanocytosis although the significance of the association is unknown.53,54 Mongolian spots are clinically diagnosed by their common location, congenital or neonatal onset, and morphology, and a biopsy is most often not needed. Other blue lesions include Nevus of Ito, Nevus of Ota, and congenital blue nevus. Location and persistence over time will help in differentiating these entities. A biopsy can be helpful in verifying the dermal location of melanocytes if the diagnosis is in question. Bluish hyperpigmentation of the skin along the ophthalmic and maxillary branches of the trigeminal nerve is known as Nevus of Ota (Fig. 24.4). These patches can be brown, blue-gray or blue-black, gray or purple and have a speckled or mottled appearance with poorly-demarcated borders. Nodular areas similar to blue nevi can be present.58–61 Nevus of Ota often affects one side of the face and bilateral involvement is described in only 5–10% of patients.62,58,63 Distribution of Nevus of Ota most commonly involves the ophthalmic and maxillary branches with involvement of all three branches of the trigeminal nerve being the least common distribution.41,60,63 This condition is more commonly seen in Asians and has a female preponderance. They may be present at birth or appear within the first year of life, or later between the ages of 11–20 years; mid-childhood onset is rare. Spontaneous resolution does not occur as seen in Mongolian spots. Pathology of cutaneous lesions shows a greater concentration of elongated, dendritic dermal melanocytes scattered within the collagen bundles in the upper dermis (compared with Mongolian spots). In addition to cutaneous involvement, Nevus of Ota involving the ocular sclera can be seen in two-thirds of patients.64 Pigmentation of the sclera, conjunctiva, cornea, iris and choroid, and less commonly, the optic nerve, retrobulbar fat, orbital periosteum and extraocular muscles can occur. Oral mucosal pigmentation and pigmentation of the tympanic membrane can occur as well. Most Nevi of Ota pose a potential cosmetic concern but few may be associated with complications. Open-angle glaucoma occurs in approximately 10% of patients due to proliferation of melanocytes in the anterior chamber angle.65,66 The most concerning complication is the development of melanoma within pigmented areas. Malignant degeneration has been reported and may include the eye (choroid, iris, or orbit), skin, and brain.67–69 Sturge–Weber syndrome, Klippel–Trenaunay syndrome, neurofibromatosis, inflammatory vascular disease, cerebral arteriovenous malformation, multiple hemangiomas, and spinocerebellar degeneration have been described occurring in association with Nevus of Ota.70–75 Rare occurrences of ipsilateral hearing loss with Nevus of Ota have been reported.76,77 Nevus of Ota occurring with nevus flammeus is seen in variants of phakomatosis pigmentovascularis. Leptomeningeal melanosis and Nevus of Ota can occur concurrently as well.68,69,75,78,79 Nevus of Ota tends to increase in size and color intensity over time. An ophthalmologic examination is recommended for those with involvement of periorbital skin, and yearly examinations for those with ocular pigmentation. Dermatologic follow-up is recommended periodically and removal of stable lesions for melanoma prophylaxis is unnecessary since the risk is quite low.58 Opaque make-up may be an option as a means of providing cosmetic improvement. Laser surgery with Q-switched devices (ruby, Nd : YAG or alexandrite) has been shown to reduce the coloration safely and remains the treatment of choice.80–84 The differential diagnosis of Nevus of Ota in a neonate includes a CALM, speckled lentiginous nevus, congenital blue nevus and ochronosis. The Nevus of Ito is an analogous lesion to Nevus of Ota with a distribution over the posterior supraclavicular and lateral cutaneous branchial nerves. These blue-gray poorly-demarcated patches may present over the shoulder, supraclavicular neck, upper arm, scapular and deltoid areas (Fig. 24.5). Bilateral nevus of Ito has been reported in a patient with a speckled lentiginous nevus,85 and bilateral Nevus of Ota with a unilateral Nevus of Ito has been noted.86 Cutaneous melanoma and malignant cellular blue nevus have been rarely reported in Nevus of Ito and extracutaneous findings are not seen.87–89 Blue nevi are dermal melanocytomas that represent benign proliferations of pigment producing dendritic dermal melanocytes (Fig. 24.6). Dermoscopy of common blue nevi reveals uniform blue coloration. Blue nevi are rarely seen at birth, but congenital forms have been described.90–92 Most are acquired, with onset in childhood or adolescence, with predilection for the scalp and face, dorsal aspects of the distal extremities and sacral area. Three types of blue nevi have been described: (1) cellular blue nevus, (2) common blue nevus, and (3) the combined nevus. Cellular blue nevi seen at birth are rare and have a predilection for the scalp, sacrococcygeal area and buttocks. They present with blue-gray or blue-black nodules or plaques with a smooth or irregular surface texture. Although most congenital cellular blue nevi are small in size (1–3 cm), larger and ‘giant’ lesions can be seen, most commonly on the scalp.93 Melanoma has been described particularly in larger lesions on the scalp.92–104 Multiple satellite blue nevi in association with a large congenital cellular blue nevus have also been reported.93,105 Other related variants of congenital blue nevi include the pauci-melanotic cellular blue nevi, epithelioid blue nevi (also known as pigmented epithelioid melanocytoma), congenital plaque-type blue nevi and neurocristic hamartomas. Multiple blue nevi may be seen in Carney complex (see below) or without associated abnormalities. A rare, congenital, plaque-like type of blue nevus, showing a circumscribed area with numerous macules and papules has been described.106 Blue nevi may arise within speckled lentiginous nevi as well as within other dermal melanocytoses such as Nevus of Ota.58,107,108 Blue-black nodules, papules and plaques seen at birth or infancy are suggestive of blue nevi. A biopsy may be needed to confirm the diagnosis and differentiate lesions from melanoma. Common blue nevi will show pigmented dendritic melanocytes singly or in small aggregations in the reticular dermis, surrounded by thickened collagen. Histopathology of cellular blue nevi, in contrast, shows cellular islands of large spindle-shaped or epithelioid cells with little or no melanin, in addition to pigmented dermal dendritic melanocytes. Lesions may penetrate deep into the subcutaneous fat.42 Infants with blue nevi that are not easy to monitor due to location and color, as well as those undergoing rapid growth or symptoms (ulceration, bleeding), should be considered for prophylactic surgical excision. Clinically stable, small common blue nevi without unusual or atypical clinical or dermoscopic features may be monitored. Although malignant blue nevi are rare, new atypical features or symptoms should be evaluated histologically via biopsy or conservative excision. Mosaicism is the presence of two or more genetically distinct cell lines in a single individual, which may arise from a variety of genetic defects from separate single mutations, groups of mutations or chromosomal differences.109 Cutaneous mosaicism is associated with diverse pigmentary patterns along the lines of Blaschko or in segmental or block-like patterns, such as the pigmentary anomalies found with hypomelanosis of Ito and McCune–Albright syndrome.27,110–112 Many hyperpigmented conditions present in segmental or Blaschko-linear patterns and may be isolated skin findings or coincide with systemic manifestations (Box 24.1).113,114 A chimera refers to an individual with cells derived from two separate zygotes that can be demonstrated by molecular analysis. Human chimerism is rare. Nevoid hyperpigmentation patterns described in human chimeras have included a flag-like rectangular pattern, a pattern of rounded units (café-au-lait-like patches) and a striated pattern.115 The term ‘linear and whorled nevoid hypermelanosis’ (LWNH) is generally used to describe a sporadic disorder of hyperpigmentation in swirls and streaks following a Blaschkoid distribution. Classically, the hyperpigmentation consists of homogenous 1–5 mm macules forming a reticulated configuration and sparing the palms, soles, mucous membranes and eyes.116 Segmental, linear or swirled hyperpigmentation or a combination of these with a sharp midline demarcation is seen over the trunk and less commonly involving the extremities (Fig. 24.7).117 LWNH appears at birth or during the first few weeks to months of life without any preceding inflammatory eruption. The pigmentation may progress for the first 1–2 years of life and then stabilize, possibly becoming less prominent over time. Biopsy of the hyperpigmented macules shows basilar hyperpigmentation with no pigment incontinence or dermal melanophages.116 Systemic abnormalities seen in association with LWNH include neurologic, developmental, musculoskeletal, and cardiac abnormalities that often arise early in life;117–119 LWNH has been described as akin to hypomelanosis of Ito (HI) with cutaneous hyperpigmentation instead of hypopigmentation. No large study exists to define the incidence of systemic features in patients with LWNH. However, systemic comorbidities are described much less frequently in association with LWNH as compared with HI.119,117 LWNH is believed to result from somatic mosaicism of neuroectodermal cells deriving from neural crest, with normal and hyperpigmented skin a result of two distinct populations of cells.116,120 Chromosomal mosaicism has also been described.114,121–126 Although thought to be a sporadic disorder, rare familial occurrences of LWNH have been reported, suggesting an undescribed gene abnormality.127 LWNH is usually diagnosed clinically and should be distinguished from other conditions with Blaschkoid hyperpigmentation (Box 24.1). With diffuse skin disease, it can be difficult to deem which coloration represents the patient’s normal skin and whether the affected areas are represented by hyper- or hypopigmentation. Skin biopsy will enable differentiation from some conditions such as incontinentia pigmenti (IP) and early (macular) epidermal nevus. No specific treatment for LWNH has been described. Thorough history and physical examination should be undertaken to exclude underlying systemic associations. If other anomalies exist, blood and skin fibroblast chromosomal analysis should be considered to look for chromosomal abnormalities. Incontinentia pigmenti (IP; see also Chapter 29) is an X-linked dominant hereditary disorder found predominantly in females, manifesting with characteristic skin findings, various tooth, eye and nail abnormalities, and neurologic and immunologic impairment. Incontinentia pigmenti is due to a mutation in the IKK-gamma (NEMO) gene.128 The cutaneous findings of the IP syndrome are evident at birth or shortly thereafter and have been classically defined by four stages: (1) vesiculobullous, (2) verrucous, (3) hyperpigmented and (4) hypopigmented.129 However, all four stages may not be seen over time, or they may overlap throughout life. The first inflammatory vesiculobullous stage presents with streaks of eosinophil-filled epidermal vesicles in a geometric fashion, mainly on the extremities, followed by verrucous or lichenoid lesions, and papules and pustules within 2–6 weeks (Fig. 24.8). This stage may persist for several months. Once resolved, widely disseminated hyperpigmentation in a whirled or linear fashion arises, mainly on the trunk. The third stage generally resolves over years and may completely disappear. A fourth stage of atrophy and hypopigmentation, commonly over the lower extremities, may replace the hyperpigmented areas. This stage may be subtle and faint and can be easily overlooked in adult females.130 The differential diagnosis of the hyperpigmented stage of IP includes linear and whorled nevoid hypermelanosis, macular epidermal nevi and other disorders with Blaschkoid pigmentation. Phakomatosis pigmentovascularis (PPV) is defined by the presence of a widespread vascular malformation, in particular a capillary malformation (port-wine stain), with cutaneous pigmented lesions such as dermal melanocytosis (usually slate-gray or blue melanosis or Mongolian spot-like) or nevus spilus with various other cutaneous nevi (nevus anemicus, epidermal nevus, cutis marmorata telangiectatica) and possible extracutaneous alterations. Pigmentation in PPV is often diffuse (>50% of the skin) and persistent and does not correlate with the typical locations for Mongolian spots (Fig. 24.9).131 A classification system based upon dominant pigmentary anomalies and subdivisions with extracutaneous findings categorizes four prominent types of PPV (Table 24.3), with type II the most common. The pathogenesis of PPV has been proposed as a phenomenon called ‘twin spotting’.132 TABLE 24.3 Classification of PPV: classification is based on accompanying pigmented nevus The designation ‘a’ and ‘b’ was given historically to denote the presence or absence of systemic findings. Numerous extracutaneous abnormalities may be seen, the most common being an overlap with Sturge–Weber syndrome, as well as Klippel–Trenaunay syndrome and melanosis oculi.133–138 The differential diagnosis of PPV includes capillary malformation-macrocephaly syndrome and Beckwith–Wiedemann syndrome. Proteus syndrome may also be considered and is distinguished by the presence of overgrowth, asymmetry and gigantism. Treatment for certain birthmarks, with the aim of improving aesthetics, such as the port wine stain and dermal melanocytosis, can be accomplished with laser therapy. Phakomatosis pigmentokeratotica (PPK) describes the sporadic concurrence of a Blaschkoid epidermal nevus (organoid or sebaceous differentiation) with a cutaneous pigmentary anomaly such as nevus spilus.139 The name rightly suggests an analogy with PPV and may occur due to the twin spotting genetic mechanism, and/or multilineage somatic mosaicism. In neonates and infants, the speckled lentiginous nevus may appear as a large CALM, prior to the development of ‘speckles’ and papules. Extracutaneous findings are usually present, including neurologic abnormalities (seizures, mental deficiency, hemiparesis and muscular weakness, dysesthesias), skeletal problems (including vitamin D-resistant rickets), and ophthalmologic alterations.139,140 Basal cell carcinoma and melanoma have been reported to arise in the nevus sebaceus and nevus spilus, respectively, and emphasizes the importance for long-term surveillance in these patients.141 Naegeli–Franceschetti–Jadassohn syndrome is an autosomal dominant subtype of ectodermal dysplasia syndrome characterized by the development of brown-gray reticulated pigmentation in early childhood without preceding inflammation.142 Pigmentation is localized on the abdomen in most cases but may be periorbital or perioral, or on the neck, flexures, groin or proximal extremities in some cases. Pigmentation gradually increases over the first decade of life and begins to fade after puberty. Sweating is markedly decreased in affected individuals, dermatoglyphics are often absent, and keratoderma of the palms and soles and dental anomalies can be seen. The syndrome is due to a mutation in the KRT14 gene located on 17q11.2-q21 and is allelic to dermatopathia pigmentosa reticularis.143 Congenital curvilinear palpable hyperpigmentation is a relatively recently described clinical finding on the posterior legs (usually bilateral posterior calves) of infants (see Fig. 8.27).145 Involvement of the arms (‘mitten-line hyperpigmentation’) and heels (‘heel-line hyperpigmentation’) has also been described.146–148 The hyperpigmented to slightly erythematous plaques are often symmetric, palpable, curvilinear and semicircumferential. Macular hyperpigmentation is also common. The etiology of these lesions is unknown but trauma from tight socks or mittens has been proposed. The pigmentation resolves spontaneously over months to years. Because of the strikingly linear configuration of the lesions, this disorder can be confused with child abuse. Xeroderma pigmentosum (XP) is a rare autosomal recessive disorder resulting in clinical and cellular sensitivity to ultraviolet (UV) light due to a decreased ability to repair DNA damage. XP patients experience cutaneous and ocular photosensitivity and are at increased risk for developing malignancy and pigmentary abnormalities (Box 24.2). An infant born with XP will manifest skin changes after exposure to UV light, including sunburn after minimal sun exposure. Numerous pigmented macules (0.1–1 cm) will develop on sun-exposed skin, usually by the age of 2 years. These lesions may be brown, gray, or black (although a single lesion will be uniform in color) and may become so dense that they coalesce into larger pigmented patches. The amount of UV exposure correlates with the degree of pigmentation. Clinically, the macules resemble freckles but they are actually solar lentigines and they do not fade over time.149 Hypopigmented or achromic spots develop and are thought to represent mutated melanocytes that have lost the ability to synthesize melanin.150,151 Telangiectasias and atrophy arise with continued UV exposure as the skin undergoes actinic degeneration and these changes may be evident by the teenage years (Fig. 24.10). The skin becomes dry, scaly, and tight appearing. Cutaneous malignancies can arise in the pigmentary stage as well as the atrophic stage and include actinic keratoses, basal cell carcinoma, squamous cell carcinoma and melanoma as well as other rare skin tumors.152–154 Ocular complications are commonly seen in XP patients as the eyelid skin, cornea and conjunctiva are exposed to sunlight. Ectropion or entropion, decreased lacrimation causing exposure keratitis, corneal edema, scarring and vascularization can develop.150 Both benign and malignant neoplasms result, including squamous cell carcinoma and ocular melanoma. An increase in internal malignancies has been reported as well.155 About 20% of patients with XP will develop primary neuronal degeneration resulting in neurologic abnormalities.155–157 These include sensorineural deafness, peripheral neuropathy, microcephaly, cerebral dysfunction (low intelligence, dementia, abnormal electroencephalogram), ventricular dilation and cortical atrophy, choreoathetosis, cerebellar ataxia, dysarthria and abnormal eye movements, and spasticity.152 The age of onset and rate of progression of the neurologic involvement is variable among patients. XP has been reported worldwide and in all races.150 It can be divided into two major forms: nucleotide excision-repair pathway defects and a variant form (XP-V), in which a defect in the post-replication repair of DNA exists.152 Designation of defects into complementation groups (XPA-G and XP-V) is based on in-vitro cell fusion studies. Different races may have a single dominant complementation group and some groups are made up of only a single kindred. Clinical features are determined by the amount of sun exposure, specific nature of the mutation and complementation group, and other environmental and unknown factors. Clinical findings at birth in a neonate with XP are lacking and not evident until UV exposure and damage occurs. An infant with significant and early sun burning should prompt further evaluation into photosensitivity syndromes. In addition to XP, other photosensitivity disorders should also be considered (Table 24.4). The differential of multiple pigmented macules includes Peutz–Jeghers syndrome, Carney complex, and LEOPARD syndrome. Histologic findings of cutaneous lesions are nondiagnostic and may show signs of severe photoaging, lentigines or malignancy. Diagnosis confirmation can be done through functional testing to screen cells for abnormalities in DNA repair, but this is available on a research basis only. Commercial molecular genetic testing is available for only a limited number of subtypes and remaining genes may be tested through research laboratories. Testing for prenatal diagnosis on cultured cells from amniocentesis can be performed if a family history of XP exists. TABLE 24.4 Photosensitivity disorders presenting in the neonatal and infantile period Management of individuals with XP involves genetic counseling, strict UV exposure avoidance, protective clothing and long hairstyles, sunscreen, sunglasses, protective window coatings and vitamin D supplementation. Skin examinations by the patient/family and dermatologist should be frequent so that malignancy and pre-malignant changes can be caught and treated early. Surgery techniques typically undertaken in normal individuals with skin cancer, such as cryotherapy, simple excision, electrodissection and curettage, Mohs micrographic surgery and the use of topical chemotherapeutic agents, are appropriate. The use of high-dose oral isotretinoin and acitretin has been reported to reduce certain types of new skin cancer formation in XP patients but use is often limited by side-effects, especially in children.158,159 Topical liposome-encapsulated endonuclease has also been reported to decrease the formation of actinic keratoses and basal cell carcinomas in XP patients.160 Radiation therapy has been used successfully in treatment of aggressive skin cancers in patients with XP, as these patients tend to have a normal cellular and clinical response to ionizing radiation.161 Death may occur in individuals with XP due to cutaneous or internal malignancy or neurologic degeneration. Post-inflammatory hyperpigmentation (PIH) refers to the term used to describe brown, blue or grayish macules and patches after an exogenous or endogenous insult. PIH can be seen at birth and early on in life. Common exogenous causes in hospitalized infants include the use of tape, adhesive from monitoring leads, and mechanical trauma, which forms distinct patterns. More indistinct forms may result from endogenous inflammatory processes seen in neonates, such as seborrheic dermatitis and atopic dermatitis. In certain skin diseases such as morphea and lichen planus, PIH presents as a definitive morphological feature; however, almost all skin disease which results in inflammation can cause PIH. Those infants with darker skin types tend to be at higher risk for developing hyperpigmentation as compared to those with lighter skin. A history of contact, trauma, or previous inflammatory process may be the clue to diagnosis. Epidermal melanocytes increase in size, number and melanin production following stimuli, including certain inflammatory diseases. When biopsied, the dermoepidermal junction and basal layer are disrupted by epidermal injury and melanin passes from its usual epidermal position to the dermis, subsequently engulfed by macrophages to form melanophages.162 PIH takes time to resolve, as dermal melanin is slow to break down. The differential diagnosis of PIH tends to be one of exclusion; certain patterns due to a particular contact are suggestive. Preceding history of an inflammatory event is helpful. A Wood’s light may aid in determining the extent of pigment alteration as it accentuates epidermal melanin. In rare cases, a biopsy may be indicated and shows melanophages in the superficial dermis and basal layer along with a variable infiltrate of lymphohistiocytes around superficial blood vessels and in dermal papillae. PIH may also be seen as a part of transient neonatal pustular melanosis (see Chapter 7) but, interestingly, a biopsy will not show dermal melanophages.162 Lesions of anetoderma of prematurity have been described to display ecchymotic-like blue-brown discoloration prior to the atrophic stage of these iatrogenic lesions.163 Hyperpigmentation usually fades over time especially when pigment is epidermal. A case of a healthy infant, born with white skin to Mexican parents, was described to develop progressive black pigmentation at 15 days of age that became diffuse by the age of 21 months, involving a portion of the palms and soles, nails, and oral and ocular mucosa.164 The infant was otherwise well and developmentally normal. A skin biopsy revealed heavy hyperpigmentation of all epidermal layers and pigment-loaded histiocytes in the dermis. This condition was thought by the authors to be due to an overproduction of melanin and termed ‘universal acquired melanosis’ or ‘carbon baby’. Other similar reports in the literature occurring in families have been termed, ‘melanosis universalis hereditaria’,165 ‘melanosis diffusa congenita’,166,167 ‘universal acquired melanosis’,164 ‘familial progressive hyperpigmentation’,168 ‘familial universal melanocytosis’, or ‘diffuse melanosis’.169 It is unclear whether these cases represent the same entity or different disorders. In general, these progressive conditions tend to describe the onset of hyperpigmentation at birth or during early infancy that progresses in extent with age. Familial progressive hyperpigmentation was originally described as hyperpigmentation in a variety of variably sized dots, whirls, streaks, and patches, in contrast to universal acquired melanosis where the entire integument becomes black.164,168 Inheritance has been described in many of these similar disorders as autosomal dominant, possibly due to a heterozygous gain of function mutation in the KIT ligand gene.170 Systemic manifestations do not occur, but irregular pigmentation of nails, oral and ocular mucosa and palms and soles have be seen. Histopathology of reported cases showed epidermal hypermelanosis with dispersed and widely distributed melanosomes within keratinocytes. The clinical features as well as the histologic absence of melanin incontinence should differentiate these conditions from IP, Naegeli–Franceschetti–Jadassohn syndrome and LWNH. Poikiloderma describes the cutaneous findings of atrophy, hyper- and hypopigmentation and telangiectasias that characterize several genetic disorders (Table 24.5, Fig 24.12). This clinical finding may be present at birth, during infancy, or develop in the first few years of life. Poikiloderma may present as a later finding in xeroderma pigmentosum, connective tissue disorders, Cockayne syndrome and Faconi anemia.171 The pathogenesis and etiology of poikiloderma are unknown, but commonly associated with UV exposure. The diagnosis is made by the typical clinical appearance of the reticulated eruption of telangiectasias and hypo- and hyperpigmentation. Histologic features depend on the severity and stage and include varying degrees of epidermal atrophy with hyperkeratosis, dilated vessels, hydropic degeneration of the basal layer, pigment-laden macrophages, and a dermal band-like or perivascular infiltrate of lymphocytes.172 The prognosis is dependant on the underlying associated disorder, and prompt recognition will aid in early diagnosis, monitoring and implementation of sun-avoidance behaviors. Addison disease is adrenocorticoid deficiency resulting from dysfunction or destruction of the adrenal cortex. Glucocorticoid and mineralocorticoid production is affected. Although onset is usually in adulthood it may present earlier in congenital adrenal hyperplasia, due to a disorder of long-chain fatty acid metabolism, or polyglandular autoimmune syndromes. Hyperpigmentation of skin and mucous membranes often precedes symptoms and is due to the stimulant effect of excess ACTH (adrenocorticotrophic hormone) and MSH (melanocyte-stimulating hormone) on melanocytes.173 Pigmentation may present as diffuse tan, brown, or bronze darkening of skin surfaces, especially on sun-exposed areas, including nails and palmar creases, and blue-back patches on mucosal surfaces (including oral and anogenital mucosa). Associated signs and symptoms include weakness, weight loss, hypotension, hyponatremia, hyperkalemia, hypoglycemia and eosinophilia. ACTH-insensitivity syndrome (or adrenocortical-unresponsiveness syndrome), a congenital insensitivity to functionally normal ACTH secretion, causes diffuse hyperpigmentation in the neonatal period and may precede systemic symptoms.174,175 Histologic findings of the hyperpigmentation are nondiagnostic and include increased epidermal melanin in keratinocytes and no increase in melanocytes.
Disorders of Hyperpigmentation and Melanocytes
Introduction
Localized hyperpigmentation – tan-brown
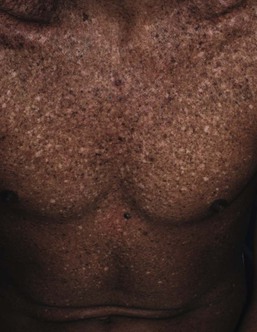
Café-au-lait macules
Strong association
Weaker associationb
Major features
Major features
Familial inherited CALMa
Multiple (≥2) generations with multiple (≥10) CALM and other NF-1 stigmata (−)
Tuberous sclerosis
Hypopigmented macules, facial angiofibromas, CNS tubers, retinal hamartomas, renal cysts, angiomyolipoma, cutaneous collagenomas, seizures, mental retardation, cardiac rhabdomyoma, periungual fibromas, subependymal giant cell astrocytomas
Neurofibromatosis type I
Skin-fold freckling, neurofibromas, Lisch nodules, neurocognitive deficits, optic glioma, skeletal dysplasia, macrocephaly
Bloom syndrome
Short stature, photosensitivity, susceptibility to malignancy, chronic lung disease, immunodeficiency, cryptorchidism
Neurofibromatosis type II
Acoustic neuromas, meningiomas, cataracts, schwannomas, neurofibromas
Ataxia-telangiectasia syndrome
Progressive cerebellar ataxia, lymphoreticular malignancy, cutaneous/ocular telangiectasia, immunodeficiency, hypogonadism
Legius syndrome
Macrocephaly, skin-fold freckling, developmental disorders
Silver–Russell syndrome
Congenital short stature and low birthweight, craniofacial and body asymmetry, limb anomalies, microcephaly
Watson syndrome
Short stature, pulmonary stenosis, low intelligence, skin-fold freckling
Bannayan–Riley– Ruvalcaba syndrome
Congenital macrosomia, megalencephaly, lipomas, intestinal polyps, vascular anomalies
Ring chromosome syndrome
Microcephaly, mental retardation, short stature, skeletal anomalies
Noonan syndrome
Facial dysmorphism, pulmonic valve stenosis, webbed neck, pectus excavatum, mental retardation, short stature, cryptorchidism, hematologic malignancies
Hereditary spinal neurofibromatosis
Skin-fold freckling, spinal root neurofibromas
Faconi anemia
Bone marrow failure, limb anomalies, mental retardation, microcephaly, predisposition to malignancy
McCune–Albright syndrome
Polyostotic fibrous dysplasia, endocrinopathies
Turner syndrome
Short stature, lymphedema, congenital heart disease, valgus deformity
LEOPARD syndrome (multiple lentigines syndrome)
Multiple lentigines, hypertelorism, pulmonic valve stenosis, EKG abnormalities, sensorineural deafness, growth retardation, genitourinary abnormalities
Multiple endocrine neoplasia syndrome type I
Parathyroid adenoma, pituitary adenoma, pancreatic islet adenoma, lipomas, gingival papules, facial angiofibromas, collagenomas
Segmental pigmentation disorder (mosaic hyperpigmentation)
Selected disorders associated with café-au-lait macules
Neurofibromatosis type 1 (NF-1)
Legius syndrome
Familial (inherited) café-au-lait macules
McCune–Albright syndrome
Disorders of dermal melanocytosis
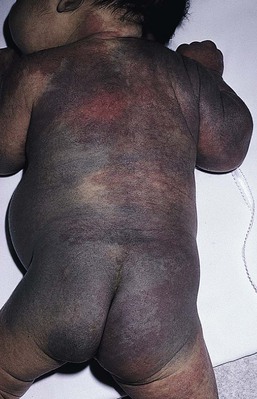
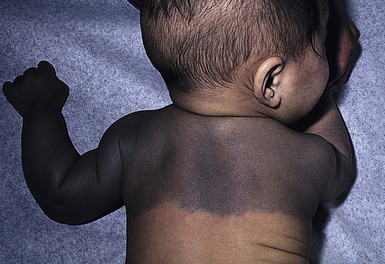
Congenital dermal melanocytosis (Mongolian spots)
Differential diagnosis.
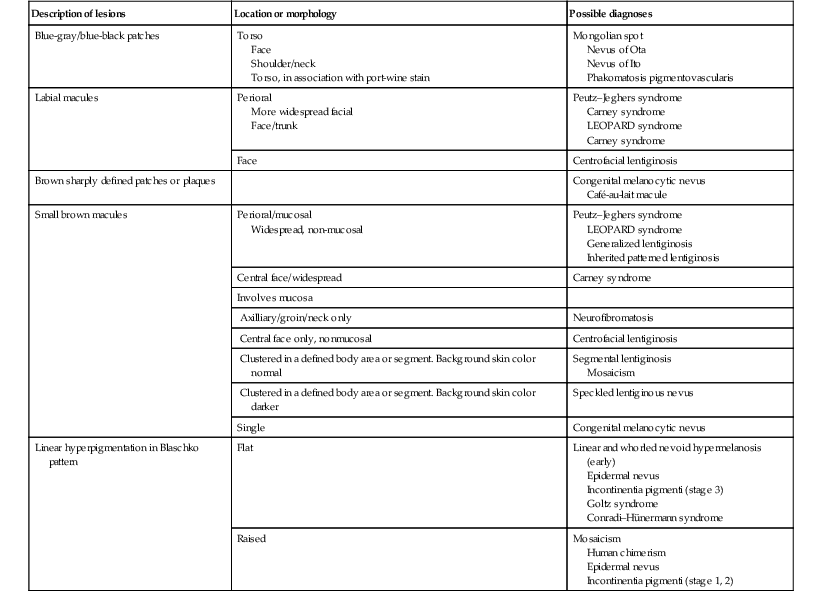
Nevus of Ota (nevus fuscoceruleus ophthalmomaxillaris, oculodermal melanocytosis)
Treatment and care.
Differential diagnosis.
Nevus of Ito (nevus fuscoceruleus acromiodeltoideus)
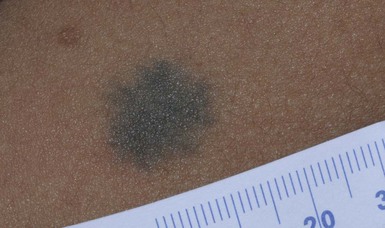
Blue nevi
Treatment and care.
Mosaic conditions and patterned dyspigmentation (whorled and segmental hyperpigmentation)
Mosaic conditions
Chimerism
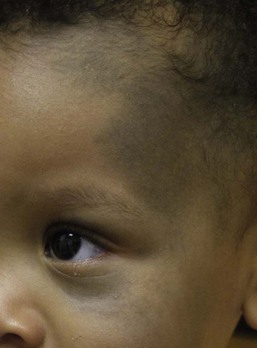
Linear and whorled nevoid hypermelanosis
Treatment and care.
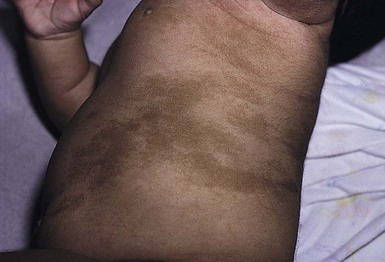
Incontinentia pigmenti
Phakomatosis pigmentovascularis
Type
Vascular malformation
Pigmentary nevus
I
Port-wine stain
Epidermal nevus
II
Port-wine stain
Dermal melanocytosis (± nevus anemicus)
III
Port-wine stain
Nevus spilus (± nevus anemicus)
IV
Port-wine stain
Dermal melanocytosis and nevus spilus (± nevus anemicus)
Phakomatosis pigmentokeratotica
Naegeli–Franceschetti–Jadassohn syndrome
Congenital curvilinear palpable hyperpigmentation (sock-line bands, sock-line hyperpigmentation)
Spotty pigmentation – diffuse
Xeroderma pigmentosum
Disorder
Defect
Findings
Cockayne syndrome
Homozygous or compound heterozygous mutation in the gene encoding the group 8 excision-repair cross-complementing protein (ERCC8) or group 6 excision-repair cross-complementing protein (ERCC6)
Cutaneous erythema with sun exposure in a butterfly distribution, atrophy, telangiectasias, neurologic impairment including ataxia and progressive deafness
Trichothiodystrophy (PIBIDS)
Mutation in helicase subunits ERCC2 and ERCC3
Photosensitivity, ichthyosis, brittle hair, impaired intelligence, decreased fertility and short stature
Congenital erythropoietic porphyria
Homozygous or compound heterozygous mutations in the uroporphyrinogen III synthase gene
Burning pain, blisters often with UV therapy for hyperbilirubinemia. Progressive scarring and disfigurement. Pink urine, red teeth, splenomegaly and anemia
Rothmund–Thomson syndrome
Compound heterozygous mutation in the DNA helicase gene RECQL4
Poikiloderma, alopecia, atrophic nails, congenital skeletal abnormalities, short stature, premature aging, and increased risk of malignant disease
Hartnup disorder
Mutations in the SLC6A19 gene
Pellagra-like light-sensitive rash, cerebellar ataxia, seizures, hypertonia, cognitive delay
Bloom syndrome
Mutations in DNA helicase RecQ protein-like-3
Poikiloderma, immunodeficiency, growth and skeletal abnormalities, predisposition to malignancy; and chromosomal instability
Treatment and care.
Post-inflammatory hyperpigmentation
Universal melanosis and progressive familial hyperpigmentation
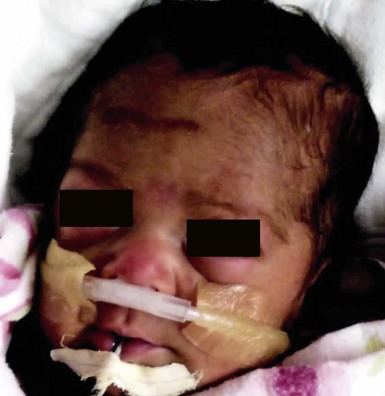
Poikiloderma
Metabolic causes
Addison disease and adrenocortical-unresponsiveness syndrome
Disorders of Hyperpigmentation and Melanocytes
24