, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
18.1 Trichorrhexis Nodosa
18.1.1 Clinical Features
The hair shaft defects of trichorrhexis nodosa can be seen in a number of genetic conditions and can also be acquired as a result of persistent trauma [1].
Trichorrhexis nodosa is characterized by brittleness and increased fragility [1]. Patients with acquired disease frequently have curly hair styled with various chemicals and straightening devices, predisposing them to the characteristic disruption of the distal hair shaft. Hair changes may improve with hairstyle modification. In congenital varieties, the hair may be normal at birth but becomes dry, dull, and more brittle during infancy and early childhood.
18.1.2 Histology
Hair mounts demonstrate the changes of trichorrhexis nodosa. There are beaded swellings along the hair shaft that are associated with focal loss of the hair cuticle (Fig. 18.1). Frayed hairs extrude out from the regions without intact cuticle giving rise to the nodular beading appearance .

Fig. 18.1
A nodular appearance resulting in hair breakage is seen in trichorrhexis nodosa (photo courtesy of Thomas Nicotri, MD)
18.1.3 Pathogenesis
The pathogenesis of trichorrhexis nodosa is not known. It can develop in association with genetic disorders, including argininosuccinic aciduria , Menkes syndrome , Netherton’s syndrome , trichohepatoenteric syndrome , and trichothiodystrophy [2]. Trichorrhexis nodosa can also be acquired due to repeated trauma to the hair shaft [3].
18.2 Trichoclasis
18.2.1 Clinical Features
Trichoclasis is a hair shaft disorder resulting from physical or chemical trauma that is not associated with an underlying defect of the cuticle, cortex, or chemical composition [4]. It is characterized by incomplete, ragged transverse fracturing of the hair shaft, demonstrated phenotypically by hair fragility [4].
18.2.2 Histology
Trichoclasis is simple transverse fracturing of the hair shaft. The hair cuticle remains intact, resulting in the hair being partially fractured but not entirely separated. The shaft itself, along with the cuticle, is structurally normal. Trichoclasis is seen in a wide range of hair disorders, and it is not a specific finding .
18.3 Loose Anagen Hair Syndrome
18.3.1 Clinical Features
Loose anagen hair syndrome has an estimated incidence of 2 cases per 1,000,000 per year [5]. The typical patients are Caucasian females with light-colored or blonde hair, who present with hair thinning or poor hair growth after 2 years of age. The mean age of diagnosis is 6 years of age, although there are reports of adult-onset hair loss [6]. Detection bias may contribute to the 6:1 female to male predominance of disease, since boys typically wear shorter hairstyles. Similarly, the microscopic features may be more difficult to appreciate in darker-haired children.
Diffuse non-scarring hair loss in loose anagen hair syndrome most often occurs at the scalp, although the eyebrows and eyelashes can be affected as well [5]. Parents frequently complain that their child’s hair does not grow long, and appears dull, fine, or unruly. Parents may note increased hair shedding with hundreds of hairs lost in a day [6]. Three phenotypes of the disorder have been described: type A with sparse hair that does not grow long; type B with diffuse unruly hair; and type C with increased hair shedding [6]. Most cases resolve spontaneously over a few years with hair growing longer, thicker and darker .
18.3.2 Histology
Hair mounts demonstrate short and twisted hair shafts lacking a surrounding cuticle and displaying distorted anagen bulbs (Fig. 18.2). Portions of the hair shafts show the expected ruffled cuticle and absent hair sheaths. There is premature keratinization of Henle’s layer that has been described as tortuous and irregularly thickened [7].

Fig. 18.2
The hair demonstrates a distorted anagen bulb and a short, twisted hair shaft lacking a cuticle in loosen anagen hair syndrome (photo courtesy of Trevor Beer, MD, Clinipath Pathology, Osborne Park, Australia)
18.3.3 Pathogenesis
Loose anagen hair syndrome is due to premature keratinization of the inner root sheath of the hair follicles, leading to impaired adhesion between the cuticle of the inner root sheath and the cuticle of the hair shaft [8, 9]. Mutations in the keratin gene K6HF, which is required for the development of the inner root sheath, have been identified in some individuals with loose anagen hair syndrome [10]. Besides K6HF, another gene K6IRS, which is specific for inner root sheath keratin formation, is also involved in disease pathogenesis [11]. This abnormal keratinization leads to impaired adhesion between the inner root sheath and the outer root sheath, resulting in short duration of the anagen hair phase and reduced hair growth [9]. These findings may explain why hair strands are loosely anchored and easily pluckable in loose anagen hair syndrome .
18.4 Trichorrhexis Invaginata
18.4.1 Clinical Features
Trichorrhexis invaginata is the pathognomonic hair shaft abnormality found in Netherton syndrome . The affected hair is sparse, short, and brittle with breakage along nodes highlighted on microscopy. The changes typically become apparent in infancy at hairs on the scalp as well as the eyebrows [1]. Hair fragility may improve with age as the hair shaft thickens [1].
18.4.2 Histology
On routine hair mounts, invagination of the hair cuticle into hair cortices can be observed [12] (Fig. 18.3). The proximal hair shaft is expanded and surrounds the distal club-shaped hair fragment. This results in an appearance of multiple small nodules or nodes [13].

Fig. 18.3
Associated with Netherton’s syndrome , linear interruptions in the hairs shafts are evidence of the invaginations seen in trichorrhexis invaginata (photo courtesy of Irina Margaritescu, MD, Bucharest, Romania)
18.4.3 Pathogenesis
Trichorrhexis invaginata (also called bamboo hair ) occurs in the setting of Netherton’s syndrome [14], and mutations in the SPINK5 (serine peptidase inhibitor, Kazal type 5) gene have been linked to this disease [15, 16]. SPINK5 encodes LEKTI (SPINK5 encoding lympho-epithelial Kazal-type inhibitor) protein, which is a secreted serine protease inhibitor expressed in stratified epithelium [17]. LEKTI is present in the stratum corneum and stratum granulosum in the skin, and it is crucial for the maintenance of epidermal barrier function [18] .
18.5 Pili Torti
18.5.1 Clinical Features
Pili torti usually presents in children before 2 years of age. It may be inherited in an autosomal dominant or autosomal recessive manner, and it can also occur sporadically [1]. Hair in pili torti is short, brittle, and sparse. Late-onset disease has been reported in older white children and adolescents with dark-colored, coarse hair at the scalp and hair fragility at the eyebrows and eyelashes. Pili torti can be seen as a feature of many genetic diseases including Bjornstad syndrome, Crandall syndrome, ectodermal dysplasia, monilethrix, and Menkes kinky hair syndrome [1].
18.5.2 Histology
On a hair mount preparation, there are hair shafts that twist upon their axes to varying degrees (Fig. 18.4). These can be 90°, 180° or 360°, and the twisting occurs with either regular or irregular frequency along the shafts. The hair shafts are somewhat flattened in appearance, and longitudinal fractures can be seen. Electron microscopy reveals irregularities in the thickness of the outer root sheath and focal vacuolization [19].
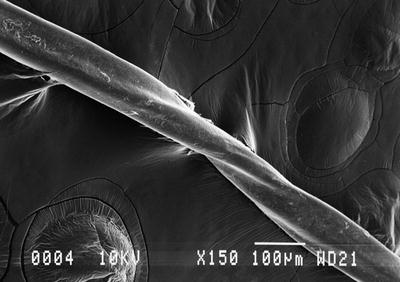
Fig. 18.4
Electron microscopy of hair shafts demonstrates a twisted appearance in pili torti (photo courtesy of Lauren Stuart, MD, Emory University, Atlanta, GA)
18.5.3 Pathogenesis
Pili torti is an autosomal dominant and acquired hair disorder in which the hair shaft is flattened at irregular intervals and twisted along its axis. The twisting of the hair strand is likely due to abnormalities in the inner root sheath, which may induce an uneven molding of the hair shaft [19]. Pili torti occurs in association with Björnstad syndrome (with sensorineural deafness) and Menkes syndrome (with early onset growth retardation and focal brain degeneration). Björnstad syndrome is caused by mutations in the BCS1L (BC1 (Ubiquinol-Cytochrome C Reductase) Synthesis-Like) gene that affect mitochondrial metabolism [20]. Menkes syndrome is an X-linked recessive disorder caused by mutations in the ATP7A (ATPase, Cu++ Transporting, Alpha Polypeptide) gene, which encodes a copper-transporting ATPase [21, 22]. Copper is an essential element in the formation of disulfide bonds in hair keratin. Therefore, defects in copper transport or metabolism may lead to abnormal keratin production, causing twisting of the hair .
18.6 Pili annulati
18.6.1 Clinical Features
Pili annulati is an uncommon disorder of hair shaft keratinization that exhibits autosomal dominant or sporadic inheritance [1]. Growth of hair is normal with normal texture and strength (Fig. 18.5). Hair at the scalp, axillae, beard, and pubis may be affected. Since hair does not demonstrate any phenotypic abnormality or fragility, treatment is not required.

Fig. 18.5
Pili annulati hair appears normal with no demonstrated abnormality (photo courtesy of MG Mercurio, MD, University of Rochester School of Medicine, Rochester, NY)
18.6.2 Histology
The diagnosis is established by the detection of alternating light and dark banding along the hair shafts. They can be seen with routine light microscopy, and the pattern of banding is reversed with reflected light. The cause of the banding is abnormalities in the hair cortex as evidenced by vacuoles within the cells [23].
18.6.3 Pathogenesis
Pili annulati is a rare autosomal dominant hair disorder. The characteristic pattern of alternating bright and dark bands of the hair strands is due to the presence of air-filled cavities along the hair cortex, which scatter and reflect the light while precluding its transmission [24]. Although the genetic cause of pili annulati is not known, the susceptibility gene locus for this hair disorder is located in the telomeric region of chromosome 12q, and may include the Frizzled gene, which encodes the transmembrane receptor for Wnt proteins [25–27].
18.7 Griscelli Syndrome
18.7.1 Clinical Features
Griscelli syndrome is a rare autosomal recessive disorder with approximately 60 confirmed cases since it was first described in 1978 [28]. There are three clinical subtypes. Type 1 is characterized by pronounced neurologic disease; type 2 by immunodeficiency with mild psychomotor disease; and type 3 has no consistent systemic abnormalities identified. There is a greater prevalence of Griscelli syndrome type 3 in individuals of Turkish and Arab ancestry, but no racial predilection has been recognized in other subtypes [29]. Griscelli syndrome is characterized by generalized pigmentary dilution with silvery, gray-colored hair at the eyebrows and scalp. Additional physical exam findings in Griscelli syndrome type 2 are pyogenic infections and cutaneous granulomas [30].
Patients with Griscelli syndrome type 1 present with severe neurologic disease at an early age, mimicking the clinical features of Elejalde syndrome (neuroectodermal melanolysosomal syndrome ), in which patients present with bronze-colored skin, silver hair, and severe neurologic impairment [28]. Griscelli syndrome type 2 has the poorest prognosis with hemophagocytic syndrome and universal fatality if bone marrow transplantation is not performed early on in the disease course [28]. Griscelli syndrome type 3 is the mildest variant of disease with no requirement for medical or surgical intervention [29].
18.7.2 Histology
In most cases, the microscopic diagnosis of Griscelli syndrome is established based upon the observation of dense clumps of pigment in an irregular distribution within the hair shaft [28, 31] (Fig. 18.6). Although not required to make the diagnosis, other histologic anomalies that have been described include giant granules in granulocytes, and enlarged and hyperpigmented melanocytes along the basal layer of the skin [28].

Fig. 18.6
Mottled pigmentation is seen throughout the hair shaft in a patient with Griscelli syndrome (photo courtesy of Anne Pham-Huy, MD, University of Ottawa, Ontario, Canada)
18.7.3 Pathogenesis
Griscelli syndrome is an autosomal recessive disorder with three clinical subtypes that are caused by distinct genetic defects. The causative mutations in type 1 Griscelli syndrome are in myosin 5A gene (Myo5A) [32]. Myo5A gene encodes an organelle motor protein that is important in neuron function [33]. A genetic mouse model that recapitulates features of type 1 is the “dilute” mouse model with loss of Myo5a [34].
Type 2 Griscelli syndrome is characterized by the same hypopigmentation of the skin and a severe immune defect of uncontrolled T lymphocyte and macrophage activation syndrome (known as hemophagocytic syndrome ), which results in severe tissue damage and organ failure [35]. Type 2 is due to mutations in Rab27a gene, which encodes a small GTPase protein involved in intracellular secretory pathways [36, 37]. There is loss of expression of Rab27a in melanocytes and cytotoxic T lymphocytes in type 2, resulting in dysfunction of lysosome-related organelles (melanosomes in melanocytes and lytic granules in cytotoxic T lymphocytes) [38, 39]. Melanocytes in Griscelli syndrome have impaired transport of melanosomes, resulting in the abnormal accumulation of mature melanosomes in the perinuclear region, in contrast to the even distribution of melanosomes throughout the cytoplasm in normal melanocytes [40]. The immune dysregulation in type 2 patients is accounted for by the requirement of Rab27a in the release of cytotoxic granules in T lymphocytes [37, 41]. A genetic mouse model with defects in melanosomes that recapitulates features of type 2 is the “ashen” mouse model with loss of Rab27a, resulting in abnormal mouse coat [42].
Type 3 Griscelli syndrome is due to mutations in the melanophilin (MLPH) gene, leading to impaired interaction of Rab27a with MLPH [43]. The same phenotype also occurs with deletion of MYO5A F-exon , an exon with a tissue-restricted expression pattern [43]. Thus, the genetic defects in the three types of Griscelli syndrome highlight the importance of Rab27a, MLPH, and Myo5A in vesicular trafficking of intracellular organelles such as the melanosome and lysosome [44].
18.8 Bubble Hair
18.8.1 Clinical Features
Bubble hair is an acquired defect in the clinical appearance and texture of the hair. It can appear at any age but is most common in adolescents and young adults. It is the result of direct heat application to wet hair during styling [45, 46]. The clinical presentation is that of tufts of wiry hair admixed with normal-appearing hair. The affected hair is dry, brittle and breaks easily. A gentle hair care technique may resolve the defect in some cases [47].
18.8.2 Histology
The hair shaft demonstrates clear bubbles in the medullary cavity that are readily visualized on direct whole mount of hair [46, 47] (Fig. 18.7). Electron microscopic examination shows loss of cortical cells and medulla at the sites of the bubbles [47].
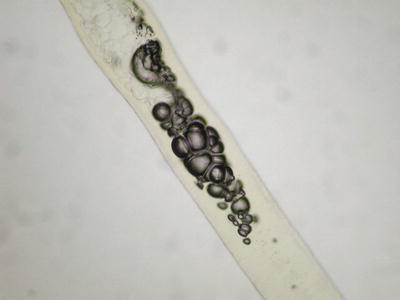
Fig. 18.7
The hair shaft contains internal cystic cavities secondary to thermal injury (photo courtesy of Catherine Antley, MD, Burlington,VT)
18.8.3 Pathogenesis
Bubble hair occurs due to heat injury to the hair by blow dryers or electric curlers used to style hair. The affected hair appears wiry with patchy loss of hair due to excessive fragility [46].
18.9 Monilethrix
18.9.1 Clinical Features
Monilethrix typically presents in early childhood [1]. The condition is characterized by autosomal dominant transmission with high penetrance but incomplete expression. The hair is “stubbled” and brittle with breaks along the characteristic elliptical nodes that are seen on microscopy. Medical therapy with topical minoxidil or oral etretinate may improve hair growth.
18.9.2 Histology
Routine light microscopy reveals a regular beading pattern within the hairs. In between the dark beads, pale constrictions are visible with fracturing in these regions (Fig. 18.8). There is a characteristic longitudinal grooving overlying the internodal pale regions, and the cuticle is absent in areas of nodes or beads [48–50].
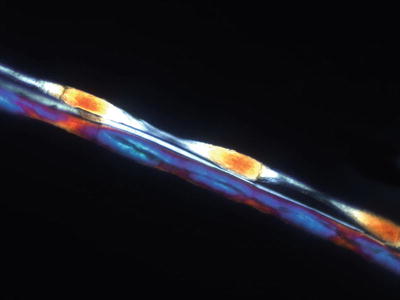
Fig. 18.8
Polarized microscopy accentuates the beaded pattern that characterizes monilethrix (Photo courtesy of Rob Law, MD, Propath Dermatopathology, Dallas, TX)
18.9.3 Pathogenesis
Monilethrix is an autosomal dominant hair disorder caused by heterozygous mutations in the hard type II (basic) keratin genes KRT81, KRT83, and KRT86 [51–53]. The most common mutations are in KRT81 and KRT86 [54]. An autosomal recessive form of monilethrix has been reported with mutations in the desmoglein 4 (DSG4) gene [55–57]. Mutated DSG4 protein predominantly accumulates in the endoplasmic reticulum (ER). Mutated DSG4 undergoes ER-associated degradation, and induces the unfolded protein response and ER stress. These biochemical changes, particularly ER stress, are important in the pathogenesis of monilethrix [56].
18.10 Trichothiodystrophy
18.10.1 Clinical Features
Trichothiodystrophy is a group of rare autosomal recessive disorders characterized by brittle hair with low sulfur content [1]. Clinical findings are vast and varied, affecting almost every organ system with eight defined primary subgroups [1, 59]. On clinical exam, patients have dry, brittle hairs that break easily. More pronounced hair shedding may occur cyclically or following infections. Hair changes typically are not amenable to therapy. Nail changes also are a prominent feature of the condition with over 60 % of affected individuals demonstrating some type of nail abnormalities, including onychodystrophy, onychoschizia, koilonychia, ridging, onychogryphosis, and yellowish discoloration [58, 59]. Forty to fifty percent of patients exhibit significant photosensitivity; however there is no increased risk for skin cancer in these patients. There is a higher incidence of premature death in affected individuals with greatest risk of mortality in children under 10 years of age, most often secondary to sepsis [58, 59].
18.10.2 Histology
The diagnosis is best established on whole hair mount and polarized microscopy. Under light polarization, alternating dark and light bands in the hair strands are evident [60] (Fig. 18.9). The hair demonstrates a regular undulating pattern, and may have some areas that appear similar to the fractures seen in trichorrhexis nodosa. The hair folds over itself creating a ribbon-like appearance [61, 62].

Fig. 18.9
Polarized light demonstrates features of banding and irregular twisting in trichothiodystrophy (photo courtesy of Trevor Beer, MD, Clinipath Pathology, Osborne Park, Australia)
18.10.3 Pathogenesis
Trichothiodystrophy has an autosomal recessive pattern of inheritance. About half of the cases exhibit skin photosensitivity and altered cellular responses to ultraviolet light. The cause of trichothiodystrophy is defects in nucleotide excision repair (NER) , which is corrects DNA damage, including that induced by ultraviolet light [63]. The causative molecular defects in the photosensitive form of trichothiodystrophy are the genes xeroderma pigmentosum complementary group B (XPB) and group D (XPD), and general transcription factor IIH subunit 5 (TTDA) [63]. XPB, XPD, and TTDA code for distinct subunits of the transcription factor IIH (TFIIH) [64]. TFIIH is a protein complex involved in both NER and transcriptional regulation [63]. XPB and XPD are ATP-dependent helicases involved in the unwinding of DNA during gene transcription and DNA repair [65, 66].
Because TFIIH functions in both gene transcription regulation and DNA damage repair, defects in the different subunits of TFIIH that affect either gene transcription or DNA repair activity provide the molecular basis for the wide variety of clinical phenotypes associated with mutations in XPB and XPD genes [67–71]. For example, mutations in the XPD subunit of TFIIH that cause defects in DNA repair mechanisms result in xeroderma pigmentosum, a distinct clinical entity of cancer-prone disorder [69].
Trichothiodystrophy also presents as a non-photosensitive form with mutations in the TTDN1 gene (TTD non-photosensitive-1) [63, 67, 72]. This form has normal response to ultraviolet light and normal levels of TFIIH. However, dermal fibroblasts from the skin of these patients overexpress matrix metalloproteinase 1 (MMP-1) that degrade collagen in the extracellular matrix [73]. The increase in MMP-1 expression is dependent on TFIIH, and leads to changes in the extracellular matrix and wound-healing properties in the skin in trichothiodystrophy .
18.11 Trichoptilosis
18.11.1 Clinical Features
Trichoptilosis or “split ends ” refers to fraying and longitudinal separation of the distal aspects of the hair shaft. It is not specific to any particular type of alopecia, but it is the end result of persistent chemical and physical trauma to the hair [74, 75]. The condition can be associated with other hair shaft abnormalities, such as trichorrhexis nodosa and trichoclasis, and it can be seen in those with congenital brittle hair [74]. In acquired cases, affected individuals may have normal appearing hair with a primary complaint of hair breakage and shedding at the scalp. Acquired trichoptilosis resolves with modification of haircare practices and products.
18.11.2 Histology
Whole mounts of hair shafts reveal fragmented and frayed ends. These changes are secondary to repeated trauma .
18.12 Melanonychia
18.12.1 Clinical Features
Melanonychia is the descriptive term for pigmentation of the nail. Longitudinal melanonychia is the most common variant of melanonychia [77]. Most cases of longitudinal melanonychia are secondary to benign melanocytic hyperplasia, primarily nevi [77]. Nail matrix nevi most commonly occur on the fingers, especially the thumb. Longitudinal melanonychia is a linear band of tan, brown, or black pigmentation extending from the nail matrix (beneath the proximal nail fold) to the distal nail plate. Even brown-black pigmentation is seen in about two-thirds of cases, with periungual pigmentation present in one-third of cases [77] (Fig. 18.10).
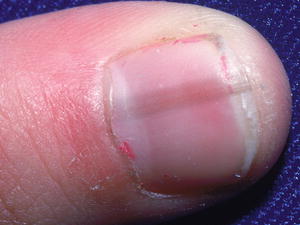
Fig. 18.10
A longitudinal pigmented streak extends across the nail in melanonychia (photo courtesy of Visual DX, Rochester, NY)
Management is dependent upon the presentation and behavior of the pigmentary band. Nail matrix melanoma in children is rare [77]. However, the focus of melanocytic hyperplasia should be removed completely if excision is pursued.
18.12.2 Histology
The clinical term melanonychia refers to pigmentation of the nail. This raises a differential diagnosis that is often easily resolved by biopsy. The histologic findings differ greatly based upon the etiology of the subungual pigment. In some cases, the pigmentation is hemosiderin secondary to subungual trauma and hemorrhage. In other cases, the pigment is melanin that resides within melanophages, and is secondary to inflammation. In these cases, it is important to completely exclude a melanocytic proliferation [78]. Fungal infection can also give rise to melanonychia. The diagnosis is established by the identification of fungal elements within the biopsy, either on routine H&E sections or on fungal stains [79]. Other cases of melanonychia are seen as part of Laugier-Hunziker syndrome, wherein increased basilar melanin pigment is present without melanocytic proliferation [80]. Lentigo simplex occurring within the subungual plate also has similar findings with many pigmented basal keratinocytes and an essentially normal number of melanocytes. In these cases, pigment incontinence may result in melanin within melanophages in the papillary dermis.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


