, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
7.1 Ehlers–Danlos Syndrome
7.1.1 Clinical Features
Ehlers–Danlos syndrome (EDS) encompasses a group of heritable connective tissue disorders characterized by joint hypermobility, dermal dysplasia, and increased fragility of vascular tissues and internal organs. Currently there are six major recognized forms of EDS (classic, hypermobility, vascular, kyphoscoliotic, arthrochalasis, and dermatosparaxis) and several rarer subtypes. Clinically, patients often present with abnormal skin texture and defective skin healing, leading to prominent scarring (Fig. 7.1). Disease prognosis depends on the type and severity [1].
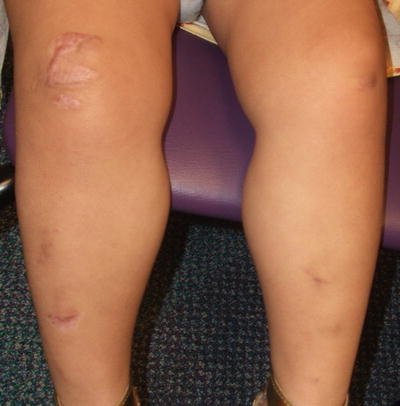
Fig. 7.1
Ehlers–Danlos Syndrome may present with atrophic “fish mouth” scars on the legs
7.1.2 Histology
Light microscopic examination contributes little to the diagnosis of Ehlers–Danlos syndrome, and it is not useful in determining the subtype of disease. Collagen bundles appear thinned, resulting in an overall thinning of the dermis (Fig. 7.2). There is increased intercellular ground substance between collagen bundles [2]. Fibroblasts with atypical morphology are present in some EDS cases [3, 4].
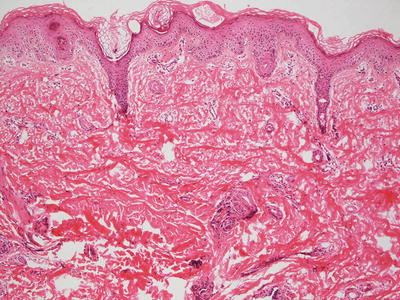
Fig. 7.2
Histologic changes are typically not seen in Ehlers–Danlos syndrome. However, collagen bundles may appear thin with overall thinning of the dermis
7.1.3 Pathogenesis
Ehlers–Danlos syndrome is a diverse group of congenital disorders characterized by musculoskeletal, skin, and blood vessel abnormalities. The classic type of EDS (type I and type II EDS ) is autosomal dominant and involves defects in type V collagen, specifically genes COL5A1 and COL5A2, as well as COL1A1 [9–11]. Type V collagen is present in many tissues, and it consists of three different α-chains encoded by the COL5A1, COL5A2, and COL5A3 genes [12]. Cysteine residues in the C-propeptide domain of α-chains are essential for disulphide bonding within the chain prior to the assembly and trimerization of collagen chains. Mutations in cysteine residues prevent proper disulphide bond formation and cause reduced levels of functional type V collagen [13, 14]. The hypermobile type of EDS (type III EDS ) is autosomal dominant, and involves defects in type I and type VI collagen, and specifically with the gene TNXB [9–11]. The vascular type of EDS (type IV EDS ) is autosomal dominant. It is the most severe form because of the risk of spontaneous vascular or visceral rupture. It involves defects in type III collagen, specifically gene COL3A1 [9–11]. There are other minor types of EDS (types VI, VIIA, VIIB, and VIIC), which are autosomal recessive and involve defects in type I collagen, particularly the genes PLOD1 and ZNF469 that are important in collagen I synthesis [9].
7.2 Marfan Syndrome
7.2.1 Clinical Features
Marfan syndrome is an autosomal dominant hereditary connective tissue disorder characterized by abnormalities primarily of the skeletal, ocular, and cardiovascular systems [15]. The main cutaneous feature of Marfan syndrome is spontaneous striae distensae of unclear etiology.
7.2.2 Histology
Skin biopsy is essentially non-diagnostic in Marfan syndrome. The epidermis is normal and the underlying dermis is remarkable only for thinned elastic tissue fibers that are not readily noticed. Ultrastructural analysis and immunofluorescence studies demonstrate decreased numbers of microfibrillar fibers [6, 16, 17]. Striae distensae are commonly seen in patients with Marfan syndrome. These lesions have the same histologic features as similar lesions occurring in patients without the underlying disease [18].
In a single case, dystrophic nails demonstrating pronounced hyperkeratosis and a sparse inflammatory infiltrate were described in a child with Marfan syndrome. The relationship between the underlying elastic tissue defect and the nail changes is not certain, and the changes may represent coexisting lichen planus [19].
7.2.3 Pathogenesis
Marfan syndrome is a connective tissue disorder with an autosomal dominant inheritance pattern. It is caused by mutations in the fibrillin-1 (FBN1) gene [20]. About 80 % of cases are hereditary, while the rest are due to de novo mutations in FBN1 [21]. Almost all of the mutations identified are unique to a particular individual or family, and there is little correlation between a specific FBN1 mutation and the clinical phenotype [22, 23]. Fibrillin-1 associates with other extracellular matrix proteins to form thread-like microfibrils , which provide elasticity and structural support to connective tissues [21, 23]. Mutations in FBN1 lead to increased degeneration of microfibrils and loss of extracellular matrix integrity.
Another key discovery in the pathogenesis of Marfan syndrome is the key role of transforming growth factor-β (TGF-β) signaling [24]. In experimental animal models of Marfan syndrome, TGF-β signaling is dysregulated in mice with loss of FBN1 gene. These mice have increased levels of TGF-β that correlated with aortic aneurysm and myxomatous changes of the atrioventricular valves , which can be prevented or reversed with neutralizing antibodies against TGF-β [25, 26]. Consistent with these experimental findings, patients with Marfan syndrome have high levels of circulating TGF-β, which can be ameliorated by treatment with drugs that have TGF-β antagonist activity. These findings indicate that TGF-β may be a therapeutic target or a biomarker for Marfan syndrome [27]. Other potential players in the pathogenesis of Marfan syndrome are ADAMTS, disintegrin and metalloproteinase with thrombospondin motif proteins. These proteins interact with fibrillin-1 and accelerate the formation of microfibrils [28, 29]. Mutations in ADAMTS proteins result in phenotypes similar those found in Marfan syndrome [28]. Thus targeting ADAMTS proteins might be a new therapeutic approach for this disease.
7.3 Cutis Laxa
7.3.1 Clinical Features
Cutis laxa, also known as elastolysis , is a rare connective tissue disorder that may be acquired or inherited. The acquired form is less common and tends to be less severe, affecting the skin only. Cutis laxa has been reported following arthropod stings and use of d-penicillamine and isoniazid [30].
The inherited forms may show autosomal dominant, recessive, or X-linked patterns. Manifestations are typically present at birth and extracutaneous involvement is common (e.g., pulmonary emphysema, umbilical/inguinal hernia, gastrointestinal and vesicourinary tract diverticula). Clinically, patients present with pendulous skin that hangs in loose folds (Fig. 7.3). There is no known effective therapy for cutis laxa.

Fig. 7.3
Cutis laxa presents as loose folds of intertriginous skin (photo courtesy of Minh Hoang, MD, Ho Chi Minh City, Vietnam)
7.3.2 Histology
It is not possible to distinguish the multiple acquired and inherited subtypes of cutis laxa on microscopic examination. Biochemical and molecular genetic testing are required to achieve this level of specificity [31, 32]. All cases of cutis laxa demonstrate absence of elastic tissue fibers in the papillary dermis and a marked diminution of fibers in the deeper portions of the dermis. Fragmented elastic tissue fibers can also be observed on routine H&E sections and on elastic tissue stains [33–36]. In some acquired cases, an antecedent inflammatory process results in enzymatic dissolution of elastic tissue fibers and remnants of the inflammation are present on biopsy [37–39]. Some authors have demonstrated subtle changes in collagen fibers in cases of cutaneous cutis laxa and suggest that the disease may represent a disorder of both elastin and collagen fibers [40].
Electron microscopic examination reveals a marked diminution of elastic tissue fibers . In cases acquired secondary to an inflammatory process, degenerating elastic tissue fibers are seen in close proximity to neutrophils [33, 38]. Vacuolated macrophages containing elastic tissue fibers are also present [34]. Marked degenerative structural changes occur within the elastic tissue fibers [41, 42].
Localized and generalized forms of cutis laxa show similar histologic findings and are only differentiated based upon clinical presentation [33, 43]. In Europe, some cases have been attributed to Borrelia burgdorferi infection [44]. Penicillamine use has been associated with alteration in cutaneous elastic tissue fibers and development of a cutis laxa-like appearance [45].
7.3.3 Pathogenesis
Cutis laxa is characterized by the loss of intact elastic fibers, resulting in loose and hypoelastic skin, most noticeably on the neck, hands, face, and groin. Cutis laxa can be inherited or acquired. Inherited forms of cutis laxa include autosomal dominant cutis laxa, autosomal recessive cutis laxa types I, IIA, and IIB, Urban–Rifkin–Davis syndrome, macrocephaly–alopecia–cutis laxa–scoliosis syndrome, arterial tortuosity syndrome, and Menkes disease/X-linked cutis laxa (XLCL) [31].
Autosomal dominant cutis laxa (ADCL) is caused by frameshift mutations in the elastin gene (ELN) as well as in the fibulin-5 gene (FBLN5) [31, 46, 47]. ELN mutations result in a missense peptide sequence in the C-terminus of tropoelastin. Mutant tropoelastin is deficient in binding to fibrillin but has enhanced self-association properties [46]. Mutant elastin leads to reduced stiffness of tissues and increased transforming growth factor (TGF)-β signaling [48].
Autosomal recessive cutis laxa-I (ARCL-I) is associated with severe systemic problems, including emphysema , arterial tortuosity and aneurysms, as well as joint laxity and muscular hypotonia [31]. Many patients with ARCL-I die from pulmonary or cardiac complications in early childhood. ARCL-I results from mutations in the genes FBLN4 and FBLN5 , which encode for fibrillin-4 and -5 [49–52]. Fibrillin-4 and -5 are required for mature elastic fiber formation and facilitate the deposition of tropoelastin onto microfibrils as well as regulating cell signaling through TGF-β [52–54].
Autosomal recessive cutis laxa-IIA , IIB , and IIIA (ARCL-IIA, IIB, and IIIA) are conditions associated with growth and developmental delay, and skeletal abnormalities [55, 56]. ARCL-IIA and B are caused by two different genes. ARCL-IIA is caused by mutations in the ATP6V0A2 gene (ATPase H+ Transporting V0 Subunit A2), which encodes a proton pump in intracellular vesicles [57, 58]. Mutations in ATP6V0A2 result in the accumulation of tropoelastin in the Golgi apparatus. ARCL-IIB is caused by mutations in PYCR1 gene (pyrroline-5-carboxylate reductase 1), which encodes a mitochondrial enzyme involved in proline metabolism [59]. ARCL-IIIA is due to mutations in the ALDH18A1 gene , which encodes for the mitochondrial enzyme Aldehyde Dehydrogenase 18 Family Member A1 that is important in the synthesis of proline, ornithine and arginine [60].
Urban-Rifkin-Davis syndrome (URDS) is a form of cutis laxa associated with severe pulmonary, gastrointestinal and urinary abnormalities. It is autosomal recessive and is caused by mutations in the latent TGF-beta binding protein 4 (LTBP4) gene [61]. The function of LTBP4 is to localize TGF-β complex to fibrillin microfibrils in the extracellular matrix [62]. Macrocephaly–alopecia–cutis laxa–scoliosis syndrome (MACS syndrome) is caused by mutations in RIN2 (Ras And Rab Interactor 2) gene, which is involved in membrane and protein trafficking in the cell [63]. Some conditions of inborn errors of metabolism, such as Menkes syndrome , are associated with cutis laxa. X-linked cutis laxa (XLCL) and Menkes syndrome are believed to be entities existing along a disease spectrum, in which patients with XLCL mainly have connective tissue problems, whereas patients with Menkes syndrome also have associated severe neurologic defects [31].
Acquired cutis laxa (ACL) is a rare disorder that often occurs in adults, and may be associated with various malignancies, infections, inflammatory dermatoses and drugs [31]. Young children can develop ACL often after a neutrophilic dermatosis , such as Sweet’s syndrome (a condition called Marshall syndrome ) [64, 65]. Patients with ACL may have low lysyl oxidase activity and α-1-antitrypsin levels, and high cathepsin G levels that could contribute to decreased skin elastin and the clinical manifestation of cutis laxa [66].
In summary, the genetic findings suggest that disruption in elastic fiber formation at various molecular levels can lead to cutis laxa conditions. Moreover, activation of TGF-β signaling is a common downstream pathway seen in these disorders [31].
7.4 Pseudoxanthoma Elasticum
7.4.1 Clinical Features
Pseudoxanthoma elasticum (PXE) is a rare inherited disorder of connective tissue that is characterized by aberrant mineralization and degeneration of elastic fibers in the soft tissues, particularly of the skin, eyes, and cardiovascular system. PXE has a prevalence of 1:50,000 in the general population, and is more common in females than males (2:1 ratio) [67]. PXE presents as small yellow papules that gradually coalesce to form larger plaques. Common locations include the neck and flexural creases. Lesions typically begin to appear during childhood or adolescence and progress into adulthood. There is no known cure for PXE, although some cutaneous lesions may be amenable to surgical excision.
7.4.2 Histology
Pseudoxanthoma elasticum is characterized by clumped and fragmented elastic tissue fibers in the dermis [68]. These abnormal elastic tissue fibers are coated with calcium deposits (Fig. 7.4). This gives a densely basophilic appearance to the elastic fibers, rendering them very apparent with routine histologic staining (Fig. 7.5). Greatly increased numbers of elastic fibers are seen, as is the thickness of the fibers themselves [69] (Fig. 7.6). Von Kossa stains highlight the calcium phosphate deposits [70]. Ultrastructural analysis reveals abnormal elastin with a granular morphology [71]. Thready material comprised of fibrinogen, collagen and glycoprotein is also detected by immunoelectron microscopy [72]. Clinically normal skin from affected individuals demonstrates calcification of normal-appearing elastic tissue fibers in the dermis [70].
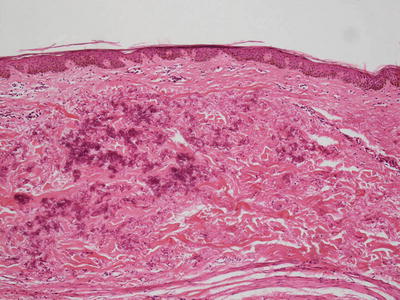
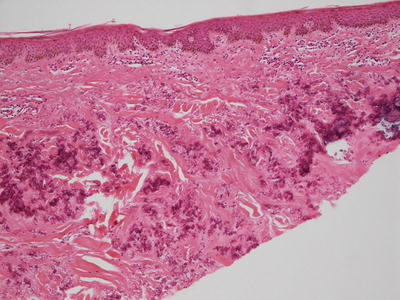

Fig. 7.4
Clumps of elastic tissue with calcification are apparent in the superficial reticular dermis in pseudoxanthoma elasticum
Fig. 7.5
The elastic tissue fibers are largely calcified and appear basophilic in pseudoxanthoma elasticum
Fig. 7.6
A greatly increased number of highly fragmented elastic tissue fibers is observed on Verhoeff–Van Gieson (VVG) stain in pseudoxanthoma elasticum
The differential diagnosis of PXE includes other conditions with disordered elastic tissue fibers including elastosis perforans serpiginosum, annular elastolytic granuloma, and wrinkling due to mid-dermal elastolysis. However, calcification is not as prominent in the other conditions. While transepidermal elimination can occur in PXE, it is not common, while it is a typical finding in elastosis perforans serpiginosum [73, 74]. Annular elastolytic granuloma has more circumscribed lesions with a palisading granulomatous response that is not seen in PXE. Wrinkling due to mid-dermal elastolysis demonstrates elastic tissue alterations confined to perifollicular regions, and the histologic changes are subtler than those seen in PXE. Long-standing treatment with penicillamine can induce changes in elastic tissue fibers that resemble those seen in PXE [75].
7.4.3 Pathogenesis
Pseudoxanthoma elasticum is a multisystem disorder with an autosomal recessive pattern of inheritance. PXE is characterized by abnormal mineralization of soft connective tissue resulting in fragmentation of elastic fibers in the skin, eyes, and cardiovascular system [76, 77]. PXE is caused by mutations in the ABCC6 gene that encodes a transmembrane ATP-binding cassette transporter protein (ABCC6/MRP6), which is normally expressed in the liver and the kidney [78–80]. The most frequent ABCC6 gene mutations are p.R1141X and g.del23-29, which account for up to 45 % of all PXE mutations [81, 82]. In experimental animal models of PXE, deletion of ABCC6 gene in mice recapitulates the genetic and morphological features of PXE with progressive mineralization of connective tissue in the affected animals [83, 84].
The mechanism of elastic fiber mineralization due to defective ABCC6 transporter is still not known. Two theories have been proposed as potential mechanisms of how ABCC6 mutations lead to pathologic mineralization in connective tissues in PXE. The “metabolic hypothesis ” states that dysfunction of ABCC6 in the liver results in deficiency of liver factor(s) required to prevent precipitation of calcium phosphate complexes and aberrant mineralization in homeostatic conditions [85, 86]. The “cell hypothesis ” states that loss of normal ABCC6 protein in the affected cells, such as skin fibroblasts, alters their biosynthetic expression profile and proliferation as well as cell–cell and cell–matrix interactions [87, 88]. Other studies have suggested that the ectopic mineralization in PXE could partially be due to vitamin K deficiency, leading to reduced levels of fully carboxylated matrix-Gla proteins (MGP) [89, 90]. Fully carboxylated MGP is a potent anti-mineralization factor expressed in connective tissues. It has been found that loss of function of the GGCX gene , which encodes a vitamin K-dependent enzyme responsible for γ-glutamyl carboxylation of Gla-proteins, results in skin lesions similar to PXE [91].
7.5 Wrinkling Due to Mid-dermal Elastolysis
7.5.1 Clinical Features
Wrinkling due to mid-dermal elastolysis (MDE) is a rare connective tissue disorder characterized by loss of elastic fibers in the mid-dermis . The cutaneous features of mid-dermal elastolysis are variable, ranging from patches of fine wrinkles to perifollicular papules and nonspecific reticular erythema [92]. There is no definitive therapy for MDE.
7.5.2 Histology
This uncommon disorder demonstrates subtle histologic changes. Elastic tissue fibers are lost in the mid-portion of the reticular dermis, sometimes with preferential loss in a perifollicular distribution [92–94]. Early cases demonstrate a mild infiltrate of lymphocytes and histiocytes. Rare multinucleated giant cells may be present and may engulf elastic tissue fibers [95]. Electron microscopic studies demonstrate degeneration of elastic tissue fibers, some of which are in macrophages [96]. While elastin is absent from regions of the dermis, the fibrillin-1 component of the fibers appears intact [97].
7.5.3 Pathogenesis
Elastin degradation in mid-dermal elastolysis is a result of increased elastase activity in the skin and subsequent phagocytosis of damaged elastic fibers by macrophages and mast cells [95, 98]. Elastase is released by inflammatory cells and skin fibroblasts. There is evidence of increased levels of matrix metalloproteinases (MMPs) and elastases , such as cathepsin G, in fibroblasts from lesional skin of patients with MDE [97, 99].
MDE may be due to an imbalance between MMPs and tissue inhibitors of MMPs (TIMPs) . The balance between activated MMPs and TIMPs determines the overall MMP proteolytic activity, and the extent of extracellular matrix degradation . Excess MMP activity can lead to pathological degradation of connective tissue, including increased elastolysis [95, 97, 100]. MDE is strongly associated with ultraviolet (UV) light exposure, and disease onset may be triggered by a phototoxic reaction [97, 101, 102]. UV light may induce the production of MMP-1 , which can lead to elastic fiber degradation in the skin [103]. Thus, a combination of increased elastase and MMP activities as well as contributing environmental factors, such as UV radiation, may underlie the pathogenesis of this disease.
7.6 Elastosis Perforans Serpiginosum
7.6.1 Clinical Features
Elastosis perforans serpiginosum (EPS) is a rare perforating dermatosis characterized by transepidermal extrusion of connective tissue elastic fibers. EPS presents as erythematous keratotic papules that coalesce into serpiginous plaques [104]. Although there is no definitive treatment for EPS, some patients have shown improvement with cryotherapy.
7.6.2 Histology
Biopsies of elastosis perforans serpiginosa demonstrate an acanthotic epidermis with a cup-shaped invagination (Fig. 7.7). Refractile eosinophilic material that is wavy and linear can be seen within the epidermis, creating a channel and extruding out of the skin (Fig. 7.8). Within the underlying dermis, clumped elastic tissue fibers appear basophilic and may be focally calcified. An elastic tissue stain is helpful in correctly identifying the fibrillar material being extruded [105] (Fig. 7.9). An inflammatory infiltrate consists of lymphocytes and occasional histiocytes [106]. A granulomatous response is present in some cases. The relationship between this condition and perforating pseudoxanthoma elasticum remains unclear [73, 107].
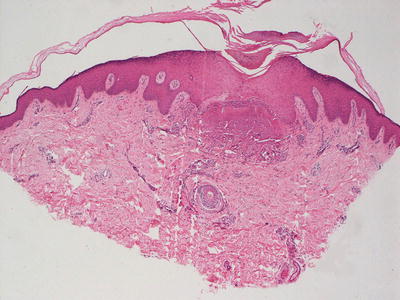
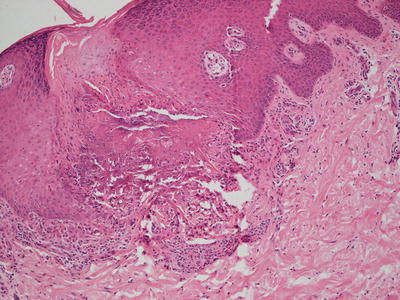
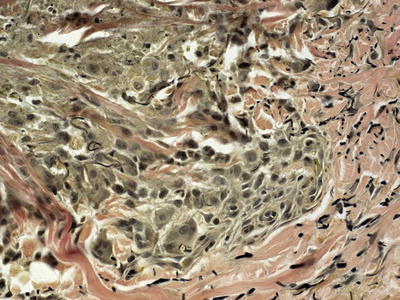
Fig. 7.7
Elastosis perforans serupignosum demonstrates an acanthotic epidermis with a central focus of disruption and underlying amorphous material in the superficial dermis
Fig. 7.8
Amorphous material is present within the papillary dermis and extends into the epidermis to the surface through a narrow channel in elastosis perforans serpiginosa
Fig. 7.9
Elastic tissue fibers in this Verhoeff-Van Gieson elastin stain are present within amorphous material that is transepidermally eliminated in elastosis perforans serpiginosa
A histologic appearance similar to EPS can be caused by long-standing treatment with penicillamine . Ultrastructural analysis D-penicillamine-induced lesions demonstrates a “lumpy-bumpy” pattern to the elastic tissue fibers , which is not seen in EPS and can be used as a distinguishing feature [108, 109]. Other perforating diseases such as Kyrle’s disease, perforating folliculitis, and reactive perforating collagenosis also enter the differential diagnosis. These entities can be differentiated in that they lack degenerating elastic fibers within the epidermis.
7.6.3 Pathogenesis
Elastosis perforans serpiginosa is a rare condition of inherited connective tissue disorders , affecting elastic tissue in the skin. EPS can be associated with systemic diseases, such as Down’s syndrome, Ehlers–Danlos syndrome, osteogenesis imperfecta, pseudoxanthoma elasticum, and Marfan syndrome [110, 111]. An acquired form of EPS has been reported in cases of long-term treatment with d-penicillamine [112]. The cause of EPS is not known, but it may be due to abnormalities induced by a variety of underlying connective tissue disorders, leading to altered elastic fibers and elimination of these fibers through the epidermis [110].
7.7 Anetoderma
7.7.1 Clinical Features
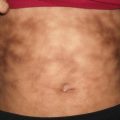
Anetoderma is a rare dermatosis characterized by focal loss of elastic tissue in the skin, resulting in circumscribed areas of loose skin. Clinically, these lesions typically present as macular depressions or pouch-like herniations of the skin (Fig. 7.10).
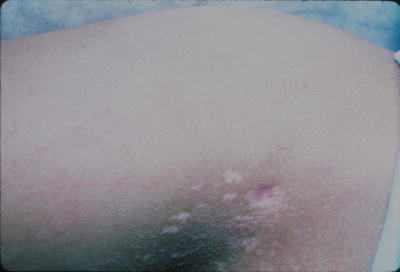
Fig. 7.10
Anetoderma presents as depigmented atrophic plaques on the extremity
Anetoderma has been classified as either primary, when there is no underlying skin disease, or secondary, when it occurs in the aftermath of another process affecting the skin (e.g., acne or varicella). Anetoderma most often affects women aged 20–40 years old [113]. However, both congenital and acquired iatrogenic forms of anetoderma have been reported in newborns. The origin of iatrogenic anetoderma in newborns has been linked to placement of medical monitoring leads on the fragile skin of premature infants [114]. Lesions are usually permanent. Some patients continue to develop new lesions for many years. Although penicillin, aminocaproic acid, and colchicine have been reported as possible therapeutic options for anetoderma, no treatment has been found to be uniformly effective for this disorder [113].
7.7.2 Histology
The epidermis in anetoderma is unremarkable, and the changes in the dermis are quite subtle and might evade detection on superficial examination. The collagen bundles may be slightly thinned, but are often relatively normal. The dermis may have decreased thickness [115]. There is an increase in the interstitial space secondary to focal elastolysis (Fig. 7.11). In early lesions, a lymphocytic or occasionally a neutrophilic infiltrate may be present within the dermis. Histiocytes are also present. It is rare to see elastolysis on routine histologic sections [116], although ultrastructural examination may show this finding [117]. In late, established lesions, elastic fibers are sparse, disrupted or even completely absent in the mid dermis (Fig. 7.12). In the majority of cases, there is an antecedent inflammatory process that is thought to cause dermal elastolysis, but these changes may or may not be present in the biopsy of clinically apparent atrophic lesions.
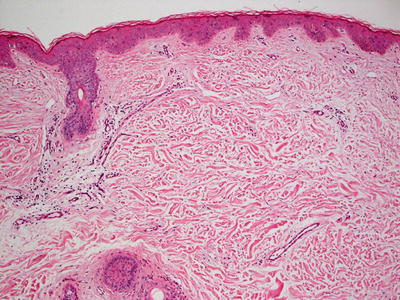
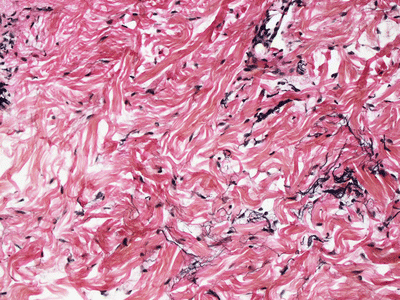
Fig. 7.11
Subtle histologic changes include slightly increased space between collagen bundles in the superficial reticular dermis in anetoderma
Fig. 7.12
Anetoderma is characterized by elastolysis and decreased elastic tissue fibers in the superficial reticular dermis as highlighted by Verhoeff-Van Gieson stain
7.7.3 Pathogenesis
Anetoderma may be caused by an imbalance between elastic fiber production and its degradation due to dysregulated production and activation of elastolytic enzymes as well as loss of elastase inhibitors. In support of this hypothesis, there is evidence of increased expression of gelatinases in skin explants from patients with anetoderma [118]. Analysis of the expression of matrix metalloproteinases (MMPs) in anetoderma skin showed increased levels of MMPs that degrade elastin as well as decreased levels of tissue inhibitor of MMPs (TIMPs) [119]. Electron microscopic studies have shown phagocytosed fragmented elastic fibers within macrophages, further suggesting an immunological mechanism in elastic fiber destruction in this disorder [117]. Immune complex deposits have been reported in the dermis and blood vessels, and granular deposits of immunoglobulin M and C3 have been found at the epidermal junction in lesional skin of anetoderma patients [120, 121].
There is a substantial body of evidence that associates anetoderma with anti-phospholipid antibodies and other serological markers of autoimmunity [122–125]. Patients with systemic lupus erythematosus and anti-phospholipid antibodies have a high risk of developing anetoderma [126, 127]. Anetoderma may be due to autoimmune-mediated destruction of elastic fibers associated with anti-phospholipid antibodies and possible shared epitopes between elastic fibers and phospholipids. Anetoderma has also been linked with other immunological diseases, such as Graves’ disease, systemic sclerosis, and autoimmune hemolytic anemia, thus supporting the clinical association of anetoderma with autoimmunity [128, 129].
7.8 Aplasia Cutis Congenita
7.8.1 Clinical Features
Aplasia cutis congenita is a rare disorder of connective tissue characterized by partial or complete absence of the skin. It may be localized or widespread. The most common site is the scalp, although it may occur anywhere on the body. Lesions appear as well-defined open wounds or translucent membranous areas. Aplasia cutis congenita typically heals with scarring (Fig. 7.13). However, large and deep defects may require skin grafting to achieve complete closure [130].
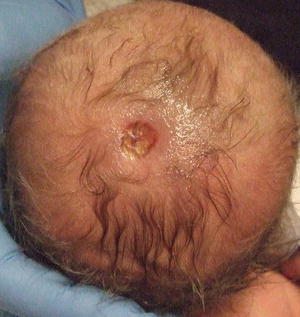
Fig. 7.13
Aplasia cutis congenita presents as a well-demarcated, round ulcer on the scalp of a neonate
7.8.2 Histology
These lesions in aplasia cutis congenita are rarely biopsied at the time of birth, at which point histologic changes would be those of ulceration and inflammation. At the usual time of biopsy, histologic sections demonstrate an atrophic epidermis with flattening of rete ridges (Fig. 7.14). The dermis has scar formation with collagen arranged linearly beneath the epidermis. The dermis is thin and lacks cutaneous appendages (Fig. 7.15). The scarring often extends into and through the subcutis to the underlying bone [131]. Dermal melanocytosis has been reported as a rare additional histologic finding in patients with aplasia cutis congenita [132].
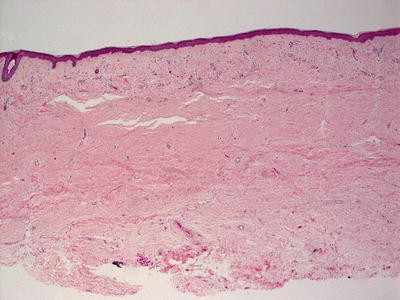
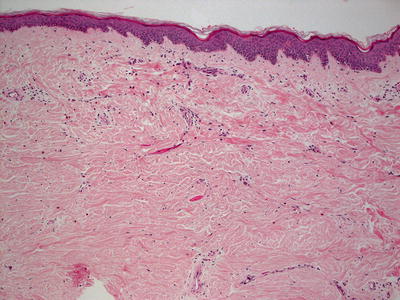
Fig. 7.14
An atrophic epidermis , lack of skin appendages and the appearance of a dermal scar characterize aplasia cutis congenita
Fig. 7.15
Loss of skin appendages, dermal fibrosis and dermal atrophy are seen in cases of aplasia cutis congenita
A rare variant of aplasia cutis congenita has been described wherein the dermis contains edematous or fibrovascular stroma. A membrane-like appearance develops into bullae. Some authors believe this subtype to be a variant of a cranial neural tube closure defect [133, 134].
The differential diagnosis of aplasia cutis congenita includes a scar secondary to other processes such as epidermolysis bullosa. Clinical history is the best way to make this distinction.
7.8.3 Pathogenesis
Genome-wide linkage analysis and exome sequencing of affected members in a family with autosomal-dominant aplasia cutis congenita identified BMS1 , a ribosomal GTPase protein, as a causative gene of this disease [135]. BMS1 is a key component in the formation of the 40S ribosomal subunit, and it is necessary for pre-ribosomal RNA processing of 18S ribosomal RNA [136, 137]. It has been shown that fibroblasts derived from aplasia cutis congenita patients with BMS1 mutation have impaired maturation of the 40S ribosomal subunit, resulting in nucleolar stress response and reduced cell proliferation rate [135]. These genetic findings link BMS1 gene and ribosome biogenesis to skin morphogenesis . In sporadic non-autosomal form of aplasia cutis congenita, many potential etiological factors have been reported, including chromosomal abnormalities, traumatic mechanism, thrombotic events, vascular alterations, and teratogens [138, 139].
7.9 Focal Dermal Hypoplasia
7.9.1 Clinical Features
Focal dermal hypoplasia (Goltz syndrome) is a rare X-linked dominant genetic disorder with a variety of cutaneous and skeletal findings as well as other organ involvement. The hallmark of focal dermal hypoplasia is connective tissue dysplasia of the skin and skeletal system (Figs. 7.16 and 7.17). Typical cutaneous features include blaschkoid linear streaks of skin atrophy and telangiectasias, fat herniations, raspberry papillomas, and dyspigmentation. Rarely, aplasia cutis congenita is also associated with the disease [140].
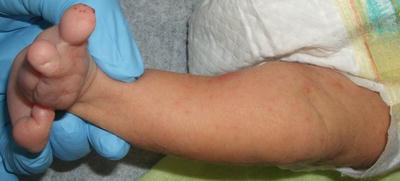
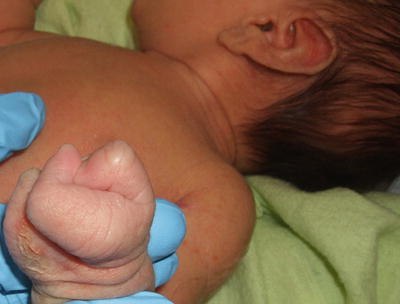
Fig. 7.16
Focal dermal hypoplasia (Goltz syndrome ) shows characteristic ectrodactyly or “lobster claw” deformity of the foot
Fig. 7.17
Focal dermal hypoplasia (Goltz syndrome) presents with ectrodactyly on the hand in the same patient with Goltz Syndrome from Fig. 7.16
7.9.2 Histology
In focal dermal hypoplasia, there is a thinned or unremarkable epidermis. The dermis is markedly diminished in thickness [141, 142] (Fig. 7.18). There are greatly reduced numbers of elastic tissue fibers [141]. Mature adipocytes are present around blood vessels and between the few residual thin collagen bundles [143, 144] (Fig. 7.19). There is an increase in number of papillary dermal blood vessels [145]. Ultrastructural analysis reveals abnormally small fibroblasts in sharply reduced numbers [141]. Adipocytes are multiloculated and appear immature [146]. The basement membrane also shows focal disruption and alteration of type IV collagen in this disorder [147]. At the periphery of long-standing atrophic lesions, there are lentigo simplex-like lesions with increased basilar melanin and slightly increased number of melanocytes [148]. Lipomas and verrucous papillomas are common findings in patients with focal dermal hypoplasia [144, 148, 149].
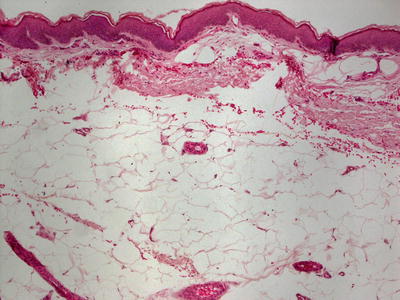
Fig. 7.18




Focal dermal hypoplasia is characterized by a normal epidermis, the absence of dermal collagen and the presence of subcutaneous fat almost adjacent to the epidermis
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
