Study type
Study design
Observational
• Cohort or panel
• Case control
• Cross-sectional
• Longitudinal
• Diagnostic
• Retrospective
Interventional
• Pilot
• Open label
• Blinded, single or double
• Randomized
• Placebo controlled
• Vehicle controlled
• Crossover
Before a new agent is studied in humans, it undergoes preclinical testing. During this phase, the primary aim of the sponsor is to use laboratory or computer-based modeling to determine the agent’s action, metabolism, and glaring toxicities. The sponsor will want to be sure the agent will not expose people to excessive risk when evaluated in small, early stage clinical studies. The agent is also evaluated for potential commercial applications. Usually, the testing is conducted using mice or rats, then possibly in larger animals.
Clinical trials involving pharmaceuticals are classified by the FDA into four phases, organized by the position in the approval process the study falls into. Before a study can begin, an investigational new drug (IND) application must be submitted to the FDA and approved. The IND effectively makes the proposed drug “legal” for the purposes of conducting research leading to FDA approval for marketing. The sponsor (applicant) for an IND may be a drug manufacturer and/or marketer, or the investigator conducting the proposed clinical investigation. The IND application will contain information about the animal pharmacology and toxicity studies, information pertaining to how the drug will be manufactured and detailed protocols for proposed early stage studies. The sponsor must wait 30 calendar days after submission of the IND before conducting any clinical trials. During this time, the FDA will review the application to be sure the research subjects will not be exposed to unreasonable risk and advise the sponsor if additional information or time is needed. Another type of IND is used for expanded access or “compassionate use.” The expanded access IND requests approval to use an investigational drug outside of clinical trials to treat patients with serious or life-threatening conditions for which no alternative treatment exists.
Once approved, human trials may begin. Phase I involves a small number of subjects (5–100) and is designed to evaluate the initial safety, side effects, and dosing range of the new drug. Pharmacology (pharmacokinetics) and metabolism of the drug are also evaluated during this phase. Phase II trials are conducted by providing the drug to a larger number or subjects to check its effectiveness and continued safety. Phase III trials encompass giving the drug to large numbers of subjects and have the purpose of collecting additional safety information, comparing it to other available treatments and confirm its effectiveness. Information collected during this phase is used to evaluate if the drug can be used safely and is the basis upon which the FDA will decide whether or not to approve the drug. Phase IV studies are conducted after approval and after the drug has been marketed. During this phase, researchers learn how the drug effects various populations and may reveal side effects associated with long-term use [7].
Inset 4.1
The following trial is of a device, measuring safety only. This was a subject and rater-blinded trial of a microneedle roller device. The sham was reproduced by applying finger pressure. Subjects were treated on the forehead, nasolabial folds, and temples. Transient erythema which was self-limited was noted with use of the device compared to the control.
Safety of a novel microneedle device applied to facial skin: a subject- and rater-blinded, sham-controlled, randomized trial. Hoesly FJ, Borovicka J, Gordon J, Nardone B, Holbrook JS, Pace N, Ibrahim O, Bolotin D, Warycha M, Kwasny M, West D, Alam M. Arch Dermatol. 2012;148(6):711–7.
Device studies are handled somewhat differently. According to FDA regulation 21 CFR 807 Subpart E [8], a 510(k) or premarket notification (PMN) is required to be made to the FDA to demonstrate that the device is safe and effective. The 510(k) process also involves an evaluation of whether the device is substantially equivalent (SE) or non substantially equivalent (NSE) to another device already approved for marketing. Submissions are reviewed and processed by the Center for Devices and Radiological Health (CDRH) within the FDA. Within the CDRH, the Office of Device Evaluation (ODE) and the Office of In Vitro Diagnostics and Radiological Health (OIR) are responsible for clearance to market in the US. Reviewers consisting of field-specific engineers, physicians, chemists, and biologists among others determine if the device is SE or NSE. New devices must be classified according to FDA regulations into one of three classes known as Class I, II, and III, determined by the device’s intended use, instructions for use, and risk-based assessment. Class I and II devices are exempt in some cases from the PMN requirements and therefore do not require a 510(k) submission. All device classes are subject to a baseline set of requirements called General Controls as defined by the Food, Drug and Cosmetic Act (FD&C). Class I devices have a long and well-understood history of use and safety profile. Examples of Class I devices include manual surgical instruments, cotton gauze for external use, and wound hydrogels. Class II devices propose more risk than Class I devices and therefore are subject to additional controls such as guidance documents, special labeling, mandatory performance standards, and post-marketing surveillance in some cases. Examples of Class II devices include sutures, some lasers, intense pulsed light devices and some RF-generating devices. Class III devices are those that support or sustain human life, are of substantial importance in preventing impairment of human health, or which present a potential, unreasonable risk of illness or injury. Class III devices are subject to the PMN process and involves demonstrating SE to another legally US marketed device. Examples of Class III devices include injectable dermal fillers, most lasers used in dermatology practice, and wound dressings that contain human cells [9]. Section 515 of the FD&C Act, Chapter V: Drugs and devices contains information on the PMA process for devices that require it [10].
Inset 4.2
Drug and Device
Combination products (prefilled syringes, MDI, transdermal patches). These are a combination of a drug prepackaged in an administration device or delivery device. They are more difficult to make, and have more regulation to deal with, but can be safer. Examples include sirolimus-eluting coronary stents by J&J (CYPHER). Dermatology drug/device combinations include pre-mixed syringes for the delivery of biologic therapies for psoriasis, and canister/drug combinations for the delivery of foam-based topical formulations. Weather a combination product is reviewed by CDRH or CDER makes a big difference. The assignment is determined by the primary mode of action. Drug eluting stents are regulated as devices, but drug eluting disks for targeted chemotherapy are regulated as drugs.
This is an example of a Combination Device and Drug Trial conducted in multiple (28) centers in the USA and Canada:
Subjects were treated with varying doses of human fetal fibroblasts in this phase 2 double blind randomized placebo-controlled trial. They had leg ulcers 2–12 cm2 in area lasting 6 weeks to 2 years. They were randomized in a 1:1:1:1:1 ratio with four escalating doses of cells or placebo. They were treated with compression bandages. All evaluators were masked during the study. The lower dose was found to be superior to vehicle in accelerating wound healing.
Kirsner RS, Marston WA, Snyder RJ, Lee TD, Cargill DI, Slade HB. Spray-applied cell therapy with human allogeneic fibroblasts and keratinocytes for the treatment of chronic venous leg ulcers: a phase 2, multicentre, double-blind, randomized, placebo-controlled trial. Lancet. 2012;380(9846):977–85.
4.2 Considerations Before Starting
So, what do you need to know before beginning a clinical trials program at your site? The first thing to know is that in order to conduct clinical research, personnel at your site must be trained in good clinical practices (GCP). GCP is an international standard for the designing, conducting, recording, and reporting of clinical trials that include the participation of human subjects. Derived from safety concerns arising in the 1960s, the World Health Organization developed guidelines to provide the public with a high level of confidence that the rights, safety, and health of trial subjects would be defended. The Declaration of Helsinki is a set of ethical principles developed by the World Medical Association (WMA) and is argued to be the gold standard in clinical trial ethics. Having undergone multiple revisions, since its inception, the Declaration of Helsinki is a living document that is in the public domain [11]. The International Conference on Harmonization (ICH) brings together regulatory authorities and the pharmaceutical industries from Europe, Japan, and the USA to promote harmonization among the groups. Additionally, there are two main government offices in the USA, the Office for Human Research Protection (OHRP) and the Office of Human Subjects Research (OHSR) that promulgate policies and procedures with respect to clinical trials. Compliance with regulations is fundamental to research practice. Free GCP training can be obtained by visiting the National Institute of Health (NIH) website at www.nih.gov [12].
Another challenge faced by prospective clinical investigators is determining and obtaining suitable training that will benefit them as they embark on a new and exciting professional opportunity. While a formal training program such as the Master of Science in Clinical Epidemiology Degree Program of the Perelman School of Medicine at the University of Pennsylvania [13] may be a good choice for someone with a desire for a career in academic medicine focused on research, those looking to add clinical research to an existing medical practice may wish to explore other options. Aside from degree-granting options, some clinical research programs offer abbreviated curricula for clinicians interested in pursuing clinical research, but may not have the time to undertake a 1 or 2-year degree [14]. Other options are presented by numerous professional organizations, which cite education as part of their mission. Examples include the Association of Clinical Research Professionals, the Society for Clinical Trials, and the Society of Clinical Research Associates which all may be found online. Some organizations offer certification programs as well as training for ancillary site staff.
Who will assist you in conducting your study? You, as the Principal Investigator (PI) are responsible for all aspects of your site’s performance in conducting a trial [15]. Certain responsibilities as defined by the protocol may be delegated to site staff such as data entry, record keeping, and scheduling of subject visits. Busy sites typically have someone other than the PI who is responsible for the day-to-day management of the site. This person is often referred to as the Study Coordinator or Clinical Research Coordinator (CRC) and must be a detail-oriented person. It is important to have identified who on your staff will serve as your CRC before going out and trying to find your first study. Subinvestigators (SubI) are other physicians at the site who conduct patient assessments, but are not as involved with the administration of the study as the PI. Depending on the size of the site as well as the number and type of studies being conducted, other personnel may be needed including a pharmacy technician, medical assistant, and office manager.
The sponsor is the pharmaceutical or device maker that is paying for and managing the trial. There will be certain personnel associated with the sponsor who will also assist your site in managing the trial. First is the study monitor or clinical research associate (CRA). CRAs visit the site on a regular basis and ensure the site is following the protocol and is responsible for ensuring proper data collection as well as documenting and reporting protocol deviations. Medical research associates (MRA) perform a role similar to that of the CRA, but are most commonly located at the sponsor’s facility. Finally, the medical monitor (MM) is a physician who has the responsibility to answer protocol and study related questions. MM’s are employed either directly by the sponsor or by a contract research organization (CRO) hired by the sponsor to manage the trial. Typically, they have study-specific knowledge and tend to have trained in the field in which the study is being conducted.
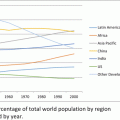
What are the physical requirements of your site? In the 1980s most clinical trials took place at universities or academic medical centers. By 2005, more than 70 % of US clinical trials were being done by nonacademic or private physicians. The number of private-sector physicians involved in studies increased from 4,000 in 1990 to 20,250 in 2010 [16




Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
