OMIM entry
Autosomal
X linked
Y linked
Mitochondrial
Totals
Phenotype described
3,814
285
4
28
4,131
Molecular basis known
Phenotype described
1,562
134
5
0
1,701
Molecular basis not known
Phenotypes with suspected Mendelian basis
1,738
115
2
0
1,855
7,114
534
11
28
7,687
In addition to hereditary etiologies, there are many environmental conditions associated with craniofacial malformations. There are numerous teratogens that can cause variable craniofacial phenotypes including ethanol, 13-cis retinoic acid (RA, Accutane), the antimetabolite methotrexate, periods of hypoxia, ionizing radiation, or hyperthermic stress [29]. The Center for Disease Control (CDC) and other groups have excellent information on teratogens that are associated with birth defects. The CDC reports that 50 % of pregnant women take four or more medications with the number of women and medications taken during pregnancy increasing over the past several decades (http://www.cdc.gov/pregnancy/features/MedUsePregnancy-keyfindings.html) [19].
Birth defects are thus etiologically diverse and continue to be a major cause of developmental defects of the craniofacial and dental complex [22]. These different conditions present a plethora of clinical manifestations that often require the support of a diverse team of clinical health-care providers to address each individual’s medical, psychosocial, and oral health-care needs. The greater the diversity of manifestations, the more likely it is that additional team members with unique expertise and knowledge will need to be involved. An example of this is the cleft lip/palate team (see Chap. 8) which will have diverse team membership to address the many different issues that accompany these conditions [32]. Whether individuals with developmental defects are cared for by a recognized team or by a community-based team of health-care providers, it is critical that effective interprofessional communication between these providers take place. Interprofessional communication will aid in obtaining a timely and accurate diagnosis and help optimize the efficiency and effectiveness of assisting the individual in the management of their health-care needs.
Developmental Defects Affecting the Dentition
Developmental defects can influence virtually every aspect of the dentition including the shape, color, size, number, composition, and the eruption and exfoliation of teeth. The human dentition begins to develop at 6 weeks in utero as the oral epithelium invaginates to initiate tooth bud formation. The molecular controls that regulate the remarkable signaling and communication between the oral ectoderm and ectomesenchyme and subsequent processes related to cell differentiation, extracellular matrix formation, mineralization, and tooth eruption are extremely complex [2, 31]. It is likely that 10,000 or more genes are involved in the making of a human tooth. Depending on the specific tooth, it takes months to years to develop a single tooth and many years to develop the primary and then permanent dentition [16]. Given the complexity and long duration required for development of the dentitions, it is not surprising that there are many different environmental insults and genetic conditions that can result in a wide variety of dental defects and manifestations [25, 27].
The exact number of developmental defects is not known, but it certainly is in the hundreds with new conditions still being described in the literature. For example, a newly described condition that affects the first permanent molar and sometimes permanent incisors (molar incisor malformation – MIM) has just recently been described for the first time [15, 34]. The first permanent molars have a thin pulp chamber and abnormal root formation, and the etiology and prevalence are unknown at this time (Fig. 1.1). There are many known hereditary and environmental conditions associated with abnormalities in tooth number (both too many and too few). The medical history, in some cases, will readily provide the cause for missing teeth, such as a child received extensive head and neck radiation and/or chemotherapy for cancer while the dentition was in its early developmental stages [8]. In other cases, the etiology will be more elusive as there may be a noncontributory medical history and no family history of missing teeth. As will be discussed in Chaps. 3 and 4, congenitally missing teeth can be associated with a syndromic condition, or it can occur as a nonsyndromic condition.
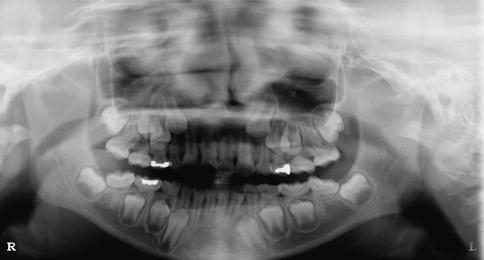
Fig. 1.1
The molar malformation that is referred to as molar incisor malformation is readily evident in this panoramic radiograph that shows the second primary and first permanent molars and maxillary permanent incisors are all affected
Teeth can form abnormally with defects in the compositions and/or structure of the enamel and/or dentin as discussed in Chaps. 6 and 7. These conditions can have significant psychosocial implications due to the importance of oral esthetics in our current society [6]. Their management often begins with eruption of the first primary tooth and continues throughout the life of the patient. Teeth may display normal crown formation but fail to erupt (Chap. 2) or may be lost prematurely (Chap. 3). Each of these different clinical manifestations provides critical diagnostic clues and will require different approaches in their management. Depending on the specific defects, a diverse dental team with expertise in a variety of areas could be needed to provide optimal oral health care over the life of the patients.
Accessing Information and Useful Databases
There are many resources that are readily available to clinicians that provide a plethora to wealth of information on conditions with craniofacial and genetic anomalies. The Internet has many different types of websites and available information. A number of very useful sites are sponsored by the National Center for Biotechnology Information (NCBI) which is a division of the National Library of Medicine. Established in 1988, NCBI creates automated systems for storing and analyzing knowledge about molecular biology, biochemistry, and genetics that can be used by researchers and health-care workers. One valuable NCBI site for clinicians is Online Mendelian Inheritance in Man (OMIM http://www.ncbi.nlm.nih.gov/omim/). This website catalogs hereditary conditions and provides information regarding clinical summaries of features, prognosis, genetic information, and direct links to references in PubMed. Each entry of a condition or gene is given a catalog number that can be used to help access the database. For example, Down syndrome is designated as OMIM # 190685. Clinicians can query this database to help access information on the clinical description of hereditary conditions and their etiology, if it is known. The OMIM database, while still incomplete, provides a wealth of information on hereditary conditions and is updated regularly. We have provided the OMIM numbers throughout this text to assist you in searching for additional information on conditions so you will be better able to diagnose and manage the oral health-care needs of your patients.
Family support groups often have extremely helpful information for both oral health-care providers and families. There are even family support groups for some of the very rare conditions. Many of these organizations have very informative websites that define the condition and have information on the current medical and dental management and information of the latest research being conducted on the diagnosis and treatment of the condition, for example, the National Foundation for Ectodermal Dysplasias, Osteogenesis Imperfecta Foundation, National Down Syndrome Society, and Cleft Palate Foundation to name just a few. Another excellent resource to get accurate information about rare conditions is the Office of Rare Disease Research sponsored by NIH (http://rarediseases.info.nih.gov/).
Taking a Family History
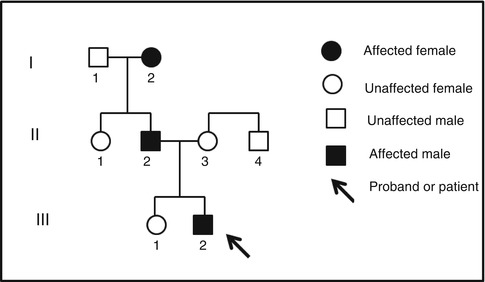
Taking a family medical history is often the critical first step in obtaining an accurate diagnosis and working with medical teams and making appropriate referrals [24]. The family medical history provides essential information for assessing whether a clinical condition may be associated with environmental causes and for assessing the potential mode of inheritance of developmental dental and craniofacial defects. Many medical organizations recommend evaluation of three generations and carefully assessing the presence of conditions in family members (e.g., American Society of Clinical Oncology, National Society of Genetic Counselors, American Medical Association). Knowledge of an individual’s family medical history is the foundation for assessing the health and risk of disease for many different conditions ranging from cancer to dental caries. Constructing a pedigree is a useful way of systematically taking a family history and recording potentially affected family members. There are numerous online resources that provide tutorials on taking a family history and constructing informative pedigrees (http://www.geneticseducation.nhs.uk/mededu/identifying-those-at-risk/taking-a-genetic-family-history). Another tool that can be used to help take a family medical history can be found on the US Department of Health and Human Services website (http://www.hhs.gov/familyhistory/portrait/). Including a question on the health history that asks whether the patient has any familial or hereditary dental or health conditions is helpful in assessing each patient. Specific questions regarding a particular phenotype (e.g., missing teeth, enamel defects, etc.) can then be asked and detailed in the history and through construction of a pedigree.
As an example, one might ask the parent of a child who has exfoliated a tooth that has not been traumatized at age 18 months whether the child’s siblings, biological parents, or biological grandparent had early loss of teeth. In this example, the parents respond that the child’s older sister has no dental problems, but the father and paternal grandmother have a similar history of early tooth loss. The most useful way to display this family history is in the form of a pedigree (Fig. 1.2). Pedigrees can be readily constructed with any computer program that can draw squares and circles. In this example, the pedigree does suggest there is a heritable risk for early tooth loss and that this trait can be transmitted from male to male and female to male suggesting an autosomal dominant mode of inheritance. This family medical history provides critical information for risk assessment and for helping direct further genetic studies that can help confirm the diagnosis.
 Developmental Defects of the Craniofacial Complex and Dentition: Scope and Challenges
Developmental Defects of the Craniofacial Complex and Dentition: Scope and Challenges
 Management of Patients with Orofacial Clefts
Management of Patients with Orofacial Clefts
 Diagnosis and Management of Defects of Enamel Development
Diagnosis and Management of Defects of Enamel Development
 Conditions Associated with Premature Exfoliation of Primary Teeth or Delayed Eruption of Permanent Teeth
Conditions Associated with Premature Exfoliation of Primary Teeth or Delayed Eruption of Permanent Teeth
 Treatment of Nonsyndromic Anomalies of Tooth Number
Treatment of Nonsyndromic Anomalies of Tooth Number
 Defects of Dentin Development
Defects of Dentin Development




Related posts:
Developmental Defects of the Craniofacial Complex and Dentition: Scope and Challenges
Management of Patients with Orofacial Clefts
Diagnosis and Management of Defects of Enamel Development
Conditions Associated with Premature Exfoliation of Primary Teeth or Delayed Eruption of Permanent Teeth
Treatment of Nonsyndromic Anomalies of Tooth Number
Defects of Dentin Development
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
