, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
With special contributions from Haider Mejbel, MD, Baylor College of Medicine.
5.1 Erythema Annulare Centrifugum
5.1.1 Clinical Features
Erythema annulare centrifugum (EAC) is a reactive dermatitis with a number of reported etiologies, including infectious antigens, drugs, neoplasm, and autoimmune diseases, although the majority of cases are idiopathic [1]. It has a peak incidence in the third and fifth decades of life with no gender predilection.
EAC presents with recurrent crops of erythematous papules that have centrifugal spread and coalesce into arcuate, annular, and polycyclic configurations (Fig. 5.1) [1]. In superficial variants of EAC, the most distinguishing feature is trailing scale along the periphery of the erythematous borders. Although the clinical course is characterized by recurrent crops of individual lesions that last for several days, the condition typically resolves spontaneously, especially after clearance of the inciting factor.
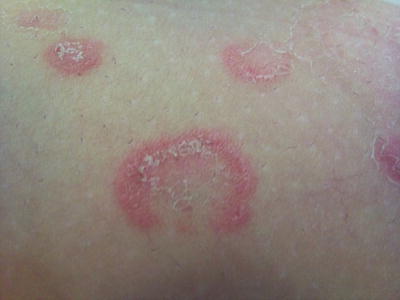
Fig. 5.1
Erythema annulare centrifugum as erythematous papules adjacent to the more classically described annular plaque with trailing scale along the leading edge of erythema (photo courtesy of Irina Margaritescu, MD, Bucharest, Romania)
5.1.2 Histology
Erythema annulare centrifugum is characterized by a dermal inflammatory infiltrate described as “tight perivascular cuffing” (Fig. 5.2). The epidermal changes are variable depending upon the biopsy site and the type of lesion biopsied. If the leading edge of the annular lesion is biopsied, the epidermis will demonstrate small foci of spongiosis, exocytosis of lymphocytes, and overlying parakeratosis. If the central portion of the lesion is biopsied, the epidermis will be entirely normal, lacking all of these changes. Papillary dermal edema may be present in some cases. The dermis has a predominantly lymphohistiocytic infiltrate that is centered around superficial blood vessels (Fig. 5.3). There is little interstitial inflammatory infiltrate, giving rise to the “tight cuffing” descriptor that is often associated with this disease. In a minority of cases, small numbers of eosinophils may be present within the dermis [2]. A “deep” variant of the disease has been described in which the inflammatory infiltrate extends to involve deeper vessels within the reticular dermis in addition to the superficial vessels. In these cases, epidermal changes are not present [3].
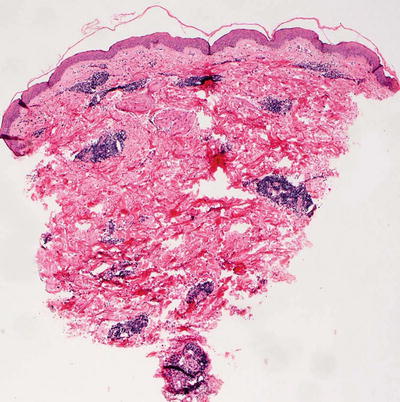
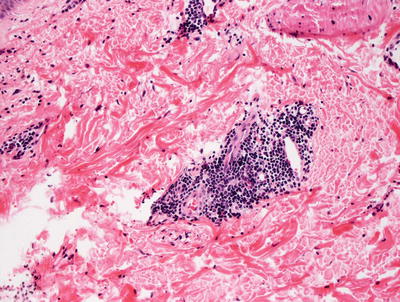
Fig. 5.2
Histologic changes in erythema annulare centrifugum include a superficial perivascular lymphocytic infiltrate that is “tightly cuffed” around vessels with little interstitial inflammation. The epidermis is unremarkable except at the edge of the lesion
Fig. 5.3
A predominantly lymphocytic infiltrate wraps tightly around dermal blood vessels in erythema annulare centrifugum
The histologic differential diagnosis of EAC is extensive because the histologic changes in this entity are not entirely specific. An arthropod bite reaction has much overlap, but the infiltrate usually contains more eosinophils between collagen bundles in the interstitium, often has a superficial punctum and extravasation of erythrocytes at the point of arthropod bite. Lupus erythematosus, especially the tumid variant, may show similar changes, but the presence of a peri-appendageal infiltrate and dermal mucin will help to distinguish lupus from EAC. Polymorphous light eruption has a very different clinical presentation and often demonstrates papillary dermal edema, a feature not associated with EAC. Some authors believe that the deep variant of EAC does not represent a distinct entity, but rather is a manifestation of one of the aforementioned processes or even a borrelial infection [4]. If the leading edge of the lesion is biopsied, the differential diagnosis is that of a mild spongiotic dermatitis and might include nummular eczema, an id reaction and atopic dermatitis .
5.1.3 Pathogenesis
The etiology of EAC is poorly understood. An immunologic basis for the disease has been proposed with the findings of IgG and C3 deposition at the basement membrane zone in lesional skin [5, 6]. EAC may represent a skin manifestation of a hypersensitivity reaction to a variety of underlying conditions, including infection, malignancy, and immunologic disorders. However, there is no increased incidence of these associations with EAC as compared with the general population [7, 8].
5.2 Pigmented Purpuric Dermatoses
5.2.1 Clinical Features
Pigmented purpuric dermatoses (PPD) are more commonly seen in adults, although reports exist of cases in children between 8 and 15 years old [9]. PPD is characterized by petechial and purpuric macules and patches, most commonly localized to the lower extremities (Fig. 5.4). The most common variants seen are described separately below.
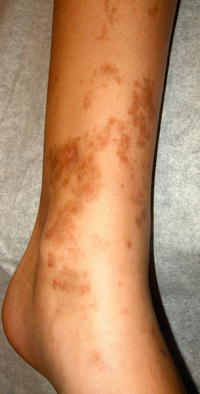
Fig. 5.4
Pigmented purpuric eruption shows coalescence of non-blanching reddish brown macules with interspersed petechiae
Progressive pigmented purpuric dermatosis / Schamberg’s disease : asymptomatic, red to slightly orange hued punctate and non-blanching macules (likened to cayenne pepper) that may coalesce into irregular shaped patches. Pruritus is an occasional complaint.
Purpura annularis telangiectoides / Majocchi’s disease : bluish-red, annular patches with associated discrete dark-red telangiectatic puncta and centrifugal spread. Occasionally, subtle atrophy is noted centrally. The site of initial involvement is usually the legs with eventual expansion to the arms.
Pigmented purpuric lichenoid dermatosis / Gougerot–Blum Syndrome : red to blue lichenoid papules that coalesce to form larger plaques on the distal legs, thighs, and trunk.
Lichen aureus: described elsewhere in this volume.
Itching and eczematoid purpura of Doucas and Kapetanakis/disseminated pruriginous angiodermatitis: pruritic generalized eruption of brown and deep red purpuric macules.
Overall, the prognosis of PPD varies. It follows a chronic and relapsing course, although spontaneous resolution is expected within months to years [10].
5.2.2 Histology
The histologic features of pigmented purpuric dermatoses vary with regards to the subtype of disease. The common features include a perivascular lymphohistiocytic infiltrate, mainly confined to the superficial vascular plexus (Fig. 5.5). Extravasation of erythrocytes and hemosiderin deposition are present within the papillary dermis, occasionally extending into the epidermis [11, 12] (Fig. 5.6). Classic features of leukocytoclastic vasculitis such as vascular wall destruction, fibrin deposition, and vascular thrombosis are not present. Eosinophils and neutrophils are not seen in this entity. In some variants of pigmented purpuric eruption, epidermal changes including lymphocyte exocytosis and spongiosis are present. In another variant, the dermal infiltrate has a lichenoid inflammatory pattern.
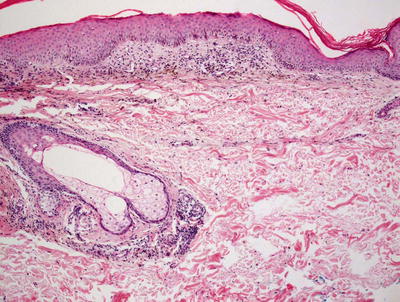
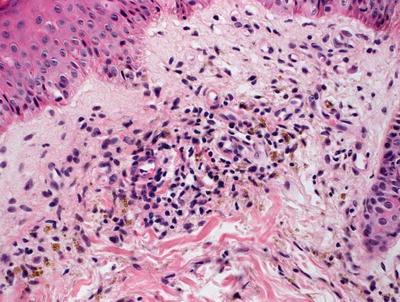
Fig. 5.5
Histologic findings in pigmented purpura are those of a superficial perivascular lymphocytic infiltrate with minimal epidermal involvement. Hemosiderin-laden macrophages are readily apparent even at low magnification
Fig. 5.6
Perivascular macrophages filled with golden brown refractile pigment granules are seen in pigmented purpuric eruptions
The histologic differential diagnosis is quite broad and depends upon the characteristics of the inflammatory infiltrate. The distinguishing feature in all cases is the presence of dermal hemosiderin, which can be highlighted on Perl’s iron stain or Prussian blue stain.
5.2.3 Pathogenesis
The etiology of PPD is not known. However, several possible contributing factors include capillary fragility, hypertension, venous stasis, and chemical exposure [13, 14]. Drugs, such as diuretics and nonsteroidal anti-inflammatory drugs, are frequent provocative factors [15].
Cell-mediated immune responses are thought to be involved in the pathogenesis of PPD. Close interactions of CD3+ and CD4+ T lymphocytes with CD1a+ dendritic cells have been identified in Schamberg disease [16]. The infiltrating immune cells in the affected areas have strong expression of leukocyte function associated (LFA)-1 antigen and ICAM-1 (Intercellular Adhesion Molecule 1) . Endothelial cells in these lesions also express ICAM-1 and ELAM-1 (E-selectin). These findings suggest that adhesion molecules may be involved in lymphocyte trafficking into the affected area and regulate lymphocyte–dendritic cell interactions. Deposits of complement factors C3 and C1q on the wall of lesional blood vessels from patients with PPD suggest that immune complexes may also play a role in the pathogenesis of these diseases [17]. The influx of inflammatory cells may predispose capillaries in the papillary dermis to dilatation and fragility with resultant rupture and hemorrhage from end capillaries [18].
5.3 Urticarial Eruptions
5.3.1 Clinical Features
Urticaria is seen in about 15–25 % of people, who will experience urticaria at some point during their lifetime [19]. Most children present with acute urticaria, and up to one-third of children will have chronic disease [19]. A causative source of urticaria can be identified in about one of five cases.
Urticaria is characterized by transient erythematous, annular, arcuate, and polycyclic papules, plaques and wheals that resolve within 24 hours of onset without residual dyschromia (Figs. 5.7 and 5.8). Lesions frequently recur in crops. Acute urticaria is defined as episodes lasting less than 6 weeks, while chronic urticaria lasts longer than 6–12 weeks [19]. Concomitant angioedema with localized swelling at the face and extremities may be seen in up to 50 % of cases of urticaria and may herald more severe episodes with risk of persistent symptoms. While most cases of acute urticaria resolve within 6 weeks, some may last longer, usually with resolution of all recurrences by 12 weeks [20]. In cases of chronic urticaria, children tend to fare better than adults, and typically are symptom-free within 3 years after disease onset.
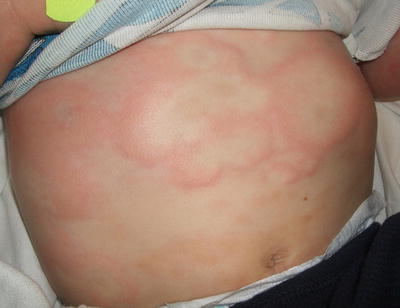
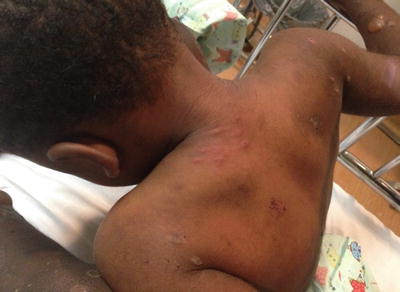
Fig. 5.7
Urticaria presents as annular, erythematous and edematous papules and plaques on the trunk of a child with preceding symptoms of an upper respiratory infection
Fig. 5.8
Papular urticaria presents as erythematous, edematous papules at exposed skin of the neck and extremities with areas of vesiculation
Giant urticaria of infancy/urticaria multiforme typically is seen in children between 4 months and 4 years of age [21]. It is most commonly reported after infectious insult, although reports exist following various antimicrobial exposures and immunizations. Urticaria multiforme is characterized by annular and arcuate, edematous and erythematous plaques with central patches of dusky-colored or ecchymotic erythema. Vesiculation and necrosis are not seen. Acral edema is a frequent feature. Patients typically have systemic symptoms suggestive of viral prodrome, including fever, malaise, cough, congestion, and diarrhea [21]. The cutaneous eruption seen in urticaria multiforme is self-limiting, usually resolving within 8–10 days without any long-term sequelae.
Papular urticaria is seen most commonly in young children and occasionally in adolescents and adults [22]. The clinical exam is characterized by pruritic, edematous and erythematous papules and wheals of varying sizes (3–10 mm) that are typically distributed at exposed skin on the extremities and face with sparing at the axillae and groin (Fig. 5.8). Lesions may persist for days to weeks, leaving residual dyschromia at previously affected sites. Symptomatic therapy is often required for active dermatitis. Recurrence following reexposure to the causative agent is the norm, although hyposensitization may develop after repeated exposures to the antigen [22].
5.3.2 Histology
In classic cases, the histologic features of urticaria are quite mild. There is a mixed inflammatory infiltrate consisting of neutrophils, eosinophils, and lymphocytes surrounding vessels of the superficial vascular plexus [23, 24] (Figs. 5.9 and 5.10). Papillary dermal edema and vascular ectasia may be present, but in many cases, are so subtle as to be undetectable following routine tissue processing [25]. It is unusual to see degranulated mast cells, and there is no significant increase in numbers of mast cells as seen on routine H&E stains [26].
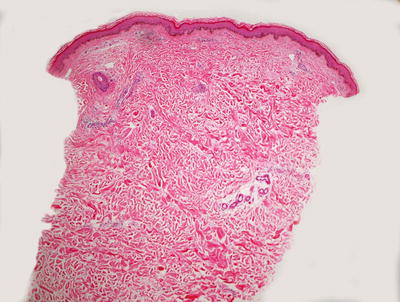
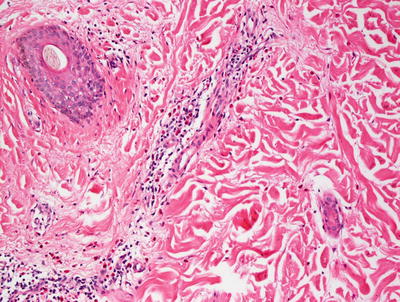
Fig. 5.9
The low magnification appearance of urticaria resembles normal skin in many cases
Fig. 5.10
Higher magnification of urticaria lesions demonstrates a mild perivascular inflammatory infiltrate consisting of eosinophils and neutrophils in addition to mast cells and lymphocytes with minimal interstitial edema. The epidermis is usually not involved
The differential diagnosis of urticaria includes a wide range of entities that demonstrate a superficial perivascular inflammatory infiltrate; however, these are usually easy to differentiate from urticaria based upon clinical history and the presence of mixed neutrophils and eosinophils in biopsies of urticaria. Urticarial vasculitis and leukocytoclastic vasculitis can be differentiated from urticaria based upon the histologic findings of overt vascular damage in leukocytoclastic vasculitis, and the clinical history and intensity of the inflammatory infiltrate in urticarial vasculitis [27]. There is some histologic overlap between urticaria and urticarial vasculitis and this distinction can be made based upon clinical definition.
Papular urticaria is an ill-defined histologic entity with features that are indistinguishable from the dermal hypersensitivity reactions in arthropod bite reactions. The overlying epidermis may show spongiosis and exocytosis in some cases, but the majority of the changes are confined to the papillary and reticular dermis (Fig. 5.11). There is a mixed inflammatory infiltrate consisting of lymphocytes, histiocytes, eosinophils, and neutrophils in varying amounts [28] (Fig. 5.12). Cases with many eosinophils may also demonstrate “flame figures” (degranulated proteins from eosinophils) [29]. The infiltrate is both perivascular and diffuse, extending between collagen bundles and deeply into the dermis and subcutis. Edema is present in varying amounts in both the papillary and reticular dermis .
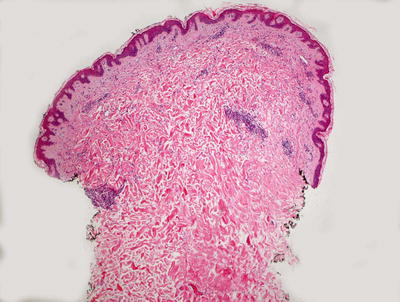
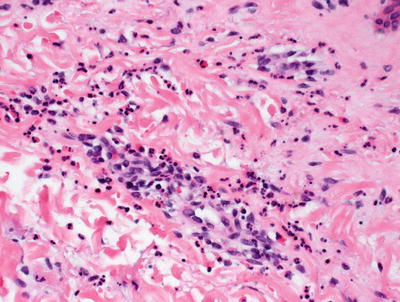
Fig. 5.11
Histologic changes in papular urticaria demonstrate a superficial perivascular mixed inflammatory infiltrate with mild dermal edema
Fig. 5.12
Neutrophils and eosinophils are present around blood vessels in papular urticaria
Urticarial vasculitis is described under leukocytoclastic vasculitis.
5.3.3 Pathogenesis
A significant proportion of cases of acute urticaria is idiopathic. When predisposing factors in children can be identified, infection is most commonly seen (identified in 49 % of affected individuals), although it may be precipitated by medication exposure (5 %) and food hypersensitivity (3 %) as well [19]. In chronic variants of urticaria, a physical etiology is more frequently cited. Granulocytes , including eosinophils, mast cells and basophils, are important in the inflammatory process of both subtypes of urticaria. The recruitment of these inflammatory cells to urticarial lesions requires the release of cytokines and chemotactic factors active on the cell types involved. The presence of intercellular adhesion molecule-1 (ICAM-1) and E-selectin in urticarial lesions suggests involvement of endothelial cell activation in active lesions [30]. Although the particular chemokines released in chronic urticaria have not been identified, there is increased plasma IL-4 in patients with chronic urticaria [31]. Lymphocytes in urticaria are both type 1 (Th1) and type 2 (Th2) T-helper cells that produce IFN-γ (by Th1 cells), and IL-4 and IL-5 (by Th2 cells) [32].
The degree of eosinophil accumulation varies among patients. However, even when eosinophils are not evident, major basic protein can be identified within lesions in the majority of cases, suggesting the prior presence of eosinophils [33]. Mast cell degranulation initiates the inflammatory process in chronic urticaria. The inflammatory infiltrate in urticaria often surrounds small venules with abundant CD4+ T lymphocytes and macrophages. Neutrophils and basophils are also present [34, 35]. Basophils have been implicated in chronic urticaria, although they are not believed to be a key activating cellular component. Instead, they may serve as bystanders that degranulate during the course of the disease [36–39].
The coagulation cascade and fibrinolysis may be involved in the pathogenesis of chronic urticaria as certain plasma factors appear to contribute to the development of skin lesions [40]. In animal models, thrombin triggers mast cell degranulation and may activate protease-activated receptor-1 on mast cells [40, 41]. Moreover, there are significantly elevated plasma levels of factor VIIa and prothrombin fragment 1 + 2 (F1 + 2) (a protein fragment that is released during the activation of prothrombin to thrombin by factor Xa) in patients with chronic urticaria as compared with normal individuals [37, 40, 41].
Studies have implicated the hormone dehydroepiandrosterone (DHEA) and its sulfated compound dehydroepiandrosterone sulfate (DHEA-S) in chronic urticaria. DHEA and DHEA-S are major androgens secreted by the adrenal glands, and they may play a role in mediating the inflammatory response in urticaria [42, 43].
Papular urticaria is thought to result from type I hypersensitivity reaction to disseminated antigen(s) deposited by an arthropod bite in a sensitized patient. This is supported by the presence of mast cells and eosinophils in the skin lesion of papular urticaria [28]. The severity of the symptoms is related to the host response to the insect salivary or contactant proteins [44]. It has been suggested that investigation of circulating immune complexes, serum complement levels, and exposure of patients to insect protein extracts may provide more insights into the immunopathogenesis of papular urticaria [28].
5.4 Annular Erythema of Infancy
5.4.1 Clinical Features
Annular erythema of infancy is a rare benign cutaneous hypersensitivity seen in healthy infants [45, 46]. Patients present with recurrent crops of asymptomatic erythematous papules that slowly enlarge and develop into arcuate and annular plaques of varying sizes on the face, trunk, and extremities. The skin eruption has a relapsing course that spontaneously resolves within months with individual lesions lasting for 2–4 weeks [46, 47]. A case of persistence into adolescence has been reported [47].
5.4.2 Histology
The literature regarding annular erythema of infancy suggests that there may be more than one process grouped into this disease category, as this is manifest by the different histologic appearances that are described [45–48]. The majority of such reports describe a dense perivascular inflammatory infiltrate consisting primarily of lymphocytes with scattered eosinophils and plasma cells [45]. In other reports of annular erythema of infancy, an interstitial infiltrate with more pronounced eosinophils has been described [46]. Additionally, in one report, a dense neutrophilic infiltrate with leukocytoclasis was noted in an individual described as having annular erythema [47]. Based upon these various histologic descriptions, it seems likely that the etiology of these clinically similar eruptions may be different.
5.5 Erythema Chronicum Migrans
5.5.1 Clinical Features
Lyme disease is the most common tick-borne disease in the USA. Erythema chronicum migrans is considered the hallmark of acute, early stage Lyme borreliosis and develops in ~80 % of affected patients [49].
Erythema chronica migrans typically is seen at the site of the tick bite within 9 days of tick attachment with a range of onset of 1–36 days [49]. It is described as an erythematous patch with centrifugal spread that may have varying degrees of erythema within the larger patch. Occasionally, there is a surrounding rim of normal pigmentation separating the initial patch from the advancing erythematous border (Fig. 5.13). Some lesions are raised but epidermal change is usually minimal. Lesions may be seen anywhere on the body, excluding the palms and soles [50]. Size of the patch is dependent on disease duration. Two-thirds of affected individuals will have concomitant systemic complaints, including fever, fatigue, malaise, arthralgia, myalgia, headache, and lymphadenopathy.
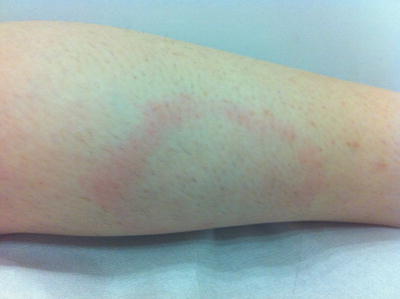
Fig. 5.13
Erythema chronicum migrans presents as an erythematous patch with centrifugal spread
Secondary erythema migrans presents with smaller and less edematous lesions distant from the original bite with the palms, soles, and mucosal surfaces typically spared [49]. Erythema usually resolves by day 8 with complete resolution of erythema chronicum migrans by 3–5 weeks [51].
5.5.2 Histology
Histologic changes of erythema chronicum migrans are those of a superficial and deep perivascular lymphohistiocytic infiltrate with no overlying epidermal changes [52–54] (Fig. 5.14). Characteristic findings include the presence of plasma cells, especially in biopsies taken from the periphery of the lesions. Some authors have described increased eosinophils in biopsies taken closer to the center of the lesions [53, 55]. A perineural infiltrate of lymphocytes and plasma cells has also been described in biopsies of erythema chronicum migrans (Fig. 5.15) [56]. An interstitial inflammatory infiltrate is also seen in some cases. In these situations, abundant histiocytes have been identified [57, 58]. Special stains for microorganisms and polymerase chain reaction testing can be used to identify borrelial microorganisms in the majority of cases.
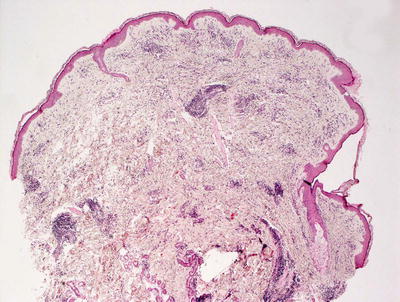
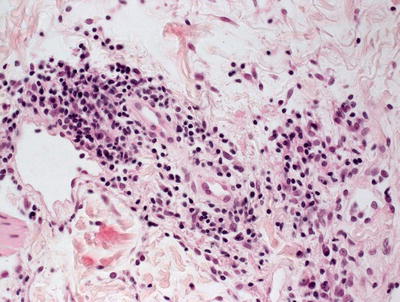
Fig. 5.14
There is a superficial and deep perivascular lymphohistiocytic infiltrate in erythema chronicum migrans. The epidermis is unremarkable
Fig. 5.15
Scattered plasma cells are present within the inflammatory infiltrate in most cases of erythema chronicum migrans
Other changes that may be seen in conjunction with erythema chronicum migrans due to cutaneous borrelial infection, but are not specific for this eruption include acrodermatitis chronic atrophicans and cutaneous lymphoid hyperplasia [57, 59].
The histologic differential diagnosis includes erythema annulare centrifugum , which does not ordinarily have plasma cells in the infiltrate. Tumid lupus erythematosus may have some similar findings, such as plasma cells, but lupus would be expected to have dermal mucin not seen in erythema chronicum migrans. Polymorphous light eruption has a similar histologic appearance, but many cases also show papillary dermal edema. The clinical presentation of each of these entities is quite different, making the distinction fairly straightforward .
5.5.3 Pathogenesis
Erythema chronicum migrans is a characteristic skin manifestation of Lyme disease caused by Borrelia burgdorferi [60]. B. burgdorferi is a pathogen that is deposited in the skin by the bite of a tick. The spirochetes can be seen on Warthin–Starry stain of biopsy of lesions, particularly in the peripheral area of the lesion [61, 62]. The disease pathology of B. burgdorferi is thought to result mainly from the host inflammatory response [63, 64].
Bacterial adhesion and immune evasion are key aspects of B. burgdorferi pathogenesis. Adhesion of the microorganisms to mammalian host cells is mediated by adhesins. A key adhesin in B. burgdorferi infection is decorin. Decorin is a small proteoglycan that is a structural component of the extracellular matrix in mammalian cells and is thought to be the extracellular matrix component to which B. burgdorferi binds [65].
Immune evasion is important in Lyme disease pathogenesis. Well-studied immune evasion strategies used by B. burgdorferi are antigenic variation and complement resistance. In antigenic variation, B. burgdorferi harbors a gene that encodes a lipoprotein called V1sE and a series of 15 silent cassettes with sequence similarly to V1sE [66–68]. Random segmented gene conversion of V1sE, in which the silent cassettes replace portions of sequences in the expressed V1sE, results in a V1sE protein with substantial antigenic variation [67, 68]. In complement resistance, B. burgdorferi has several complement regulatory factor H-binding proteins that interfere with the complement pathway by promoting inactivation of complement protein C3b [69, 70]. In addition to these immune-mediated mechanisms , the innate immune system is also important in host response to B. burgdorferi infection, specifically pathogen-associated molecular patterns (PAMPs) that are recognized by pattern recognition receptors (PRRs) expressed by mammalian immune cells [66].
5.6 Scleroderma/Morphea
5.6.1 Clinical Features
Scleroderma is a chronic connective tissue disorder. It is divided into two main categories: systemic sclerosis and localized scleroderma or morphea. Scleroderma is a rare condition with annual incidence in the pediatric population of 1 per million for systemic sclerosis, and 2 to 3 per 100,000 for localized scleroderma [71]. Fewer than 5 % of patients with systemic sclerosis have onset during childhood, while the majority of patients with localized scleroderma develop disease during childhood. In children, the mean age of onset is between 7 and 8 years old [71]. Ninety percent of children are diagnosed between 2 and 14 years old [72]. Scleroderma is more common in females, with a female to male ratio of 4:1 for systemic sclerosis and 2:1 for localized scleroderma.
Systemic sclerosis is characterized by cutaneous sclerosis with visceral involvement. There are three defined subtypes: diffuse cutaneous, limited cutaneous, and the overlap type.
In localized scleroderma, only the skin and underlying tissues are affected. Localized scleroderma is divided into five subtypes: plaque morphea, generalized morphea, bullous morphea, linear morphea, and deep morphea. Localized areas of indurated skin, often with dyspigmentation, characterize plaque morphea . Active lesions typically have a purplish halo. In the generalized form, patients have indurated plaques involving more than two body sites. Bullous morphea is characterized by the presence of bullae and erosions overlying indurated plaques. Deep morphea commonly presents as a single atrophic lesion on the upper back.
Linear morphea is the most common form of localized scleroderma seen in childhood [72]. Approximately 67 % of patients with linear morphea are diagnosed before 18 years old. Typically, a single unilateral lesion develops on the extremities, face or scalp (Figs. 5.16, 5.17 and 5.18). The underlying muscles and bones can be affected, causing growth disturbance and the potential for significant deformities.
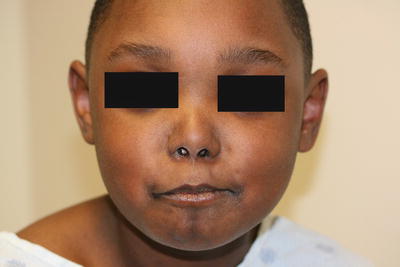
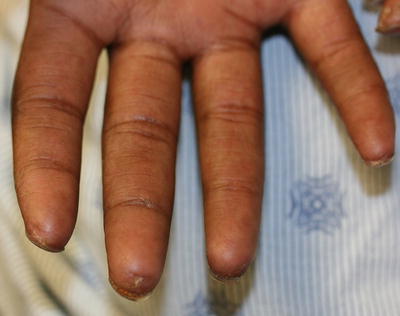
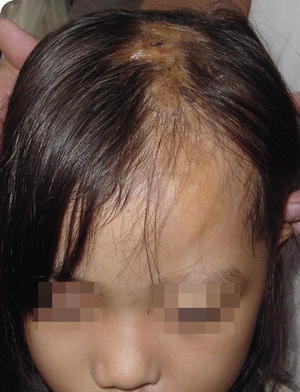
Fig. 5.16
Linear morphea presents as a dimpled, slightly hypopigmented linear plaque involving the left nasolabial fold and extending to the nasal ala and upper lip with subsequent asymmetry of the aperture of the ipsilateral nostril
Fig. 5.17
Sclerotic tightening of the digits and ulcerations at the fingertips are seen in a patient with scleroderma (photo courtesy of Minh Hoang, MD, Ho Chi Minh City, Vietna)
Fig. 5.18
Linear morphea presents as a hyperpigmented sclerotic band extending from the lateral forehead to the crown of the scalp with concomitant alopecia and hemorrhagic ulceration (photo courtesy of Minh Hoang, MD, Ho Chi Minh City, Vietnam)
Linear lesions affecting the scalp and forehead are called “en coup de sabre” due to the development of a depressed furrow resembling a blow from a saber. En coup de sabre typically is localized to the paramedian forehead with associated ocular and central nervous system (CNS) symptoms, such as headache, seizures, peripheral neuropathy, vascular malformation, and CNS vasculitis [73].
Another variant of linear scleroderma affecting the face is Parry–Romberg syndrome , also known as progressive hemifacial atrophy . In this condition, there is slow unilateral atrophy of the facial tissues, including skin, muscles, cartilage, and bones. There may be associated neurologic and ophthalmologic manifestations. In linear limb morphea, the involvement usually is unilateral with sequelae including muscle atrophy, joint contractures as well as limb length and limb circumference discrepancy [73].
The majority of patients will have softening and stabilization of disease over a 3–5-year period, although dyschromia and scarring may be permanent [74]. One-third of patients will have persistence of disease into adulthood, warranting continued clinical monitoring. Patients with linear morphea are more likely to have concomitant systemic disease than those with localized scleroderma. About 25 % of affected children have extracutaneous symptoms, often involving the musculoskeletal system [73, 74].
Prognosis depends on the extent and severity of disease. Localized lesions are often treated with topical therapy (e.g., topical steroids, calcineurin inhibitors, and vitamin D derivatives). More widespread debilitating disease is usually treated with one or more immunosuppressants. Phototherapy has also been used with some success. In general, localized forms of scleroderma will have a self-limited course. However, disease activity may persist for months to years and can be recurrent. Systemic sclerosis typically has a chronic course. Children tend to have a better long-term prognosis than adults with less likelihood of severe organ involvement [71].
5.6.2 Histology
Scleroderma and morphea cannot be distinguished based upon histologic changes alone. Regardless of the clinical presentation, the histologic changes vary with the age of the lesion biopsied. Histologic changes are largely confined to the reticular dermis. In late stage lesions, similar changes may extend to the papillary dermis and into the fibrous septae in the subcutaneous fat. Early lesions demonstrate dermal edema leading to expansion of the spaces between reticular dermal collagen bundles. There is a perivascular, peri-appendageal, and interstitial inflammatory infiltrate that consists of lymphocytes, scattered plasma cells, and eosinophils (Fig. 5.19). As lesions evolve, the edema diminishes and the space between collagen bundles becomes decreased with progressive thickening of these bundles (Fig. 5.20). The overall intensity of the inflammatory infiltrate becomes less and the inflammation is centered more on blood vessels, hair follicles, and eccrine structures. The presence of plasma cells in these foci is expected and is diagnostically useful (Fig. 5.21). Occasional germinal follicles with scattered plasma cells may be present, especially at the interface between the deep reticular dermis and the subcutaneous fat. In late stage lesions , the inflammation may be entirely absent with only dense sclerotic collagen with minimal interstitial spaces. In this phase, it is common to see fibrous tracts that replace obliterated hair follicles and eccrine ducts. Elastic tissue fibers are preserved throughout the dermis in morphea and scleroderma [75–77]. Similar changes have been described in late stages of Borrelia burgdorferi infection and the relationship between this infectious process and scleroderma remains somewhat controversial.
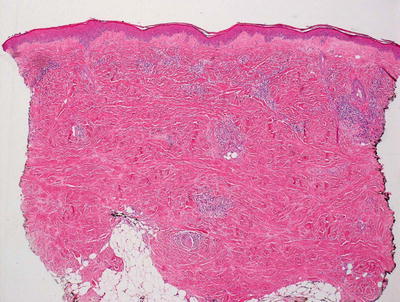
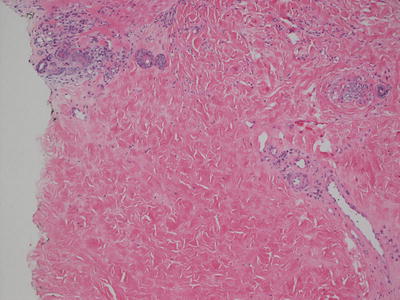
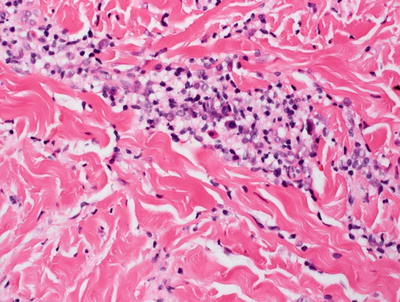
Fig. 5.19
Morphea shows a perivascular and interstitial infiltrate of lymphocytes, scattered plasma cells, and eosinophils. A “square biopsy” sign is characteristic of morphea since the dermis is distended with increased and thickened collagen bundles
Fig. 5.20
There are thickened collagen bundles, decreased interstitial spaces and minimal interstitial inflammation in morphea
Fig. 5.21
Dense sclerosis and a lympho-plasmacytic cell infiltrate are present in the deep reticular dermis in morphea
The relationship between lichen sclerosus and morphea remains unclear [78]. Certainly, in many patients with morphea , histologic changes of lichen sclerosus are present within the papillary dermis and epidermis, while the reticular dermis has the characteristic features of morphea. A rare variant of morphea has been described in which only the most superficial portion of the dermis is affected by the increased fibrosis [79].
The histologic differential diagnosis of morphea includes a dermal scar, which can be differentiated based upon the linearity of the collagen bundles and the vertical orientation and linearity of dermal blood vessels seen in a scar. Also to be considered is scleredema. In this entity, while collagen bundles may be thickened in late stage lesions, there is increased interstitial space separating the collagen bundles. The spaces are filled with mucin as highlighted with alcian blue or colloidal iron stains. Nephrogenic systemic fibrosis may have some histologic similarity, but demonstrates increased numbers of fibroblasts, which is a feature not seen in morphea and scleroderma. Tumid lupus erythematosus may have a superficial and deep perivascular and peri-appendageal inflammatory infiltrate with scattered plasma cells. However, the dermal sclerosis seen in morphea and scleroderma are not present.
5.6.3 Pathogenesis
Scleroderma and morphea are characterized by the overproduction of collagen and increased extracellular matrix deposition. It has been suggested that scleroderma is due to microvascular injuries with inflammatory cell infiltrates in the dermis and surrounding blood vessels present in early morphea, resulting in increased production of vascular cell adhesion molecule-1 (VCAM-1) , intercellular adhesion molecule-1 (ICAM-1) and E-selectin, as well as chemokines (CCL2, CCL5, and CXCL8) [80–82]. These pro-inflammatory molecules stimulate T-helper cell activation and the production of key profibrotic cytokines , such as transforming growth factor-β (TGF-β), tumor necrosis factor (TNF)-α, interferon (IFN)-α, IFN-γ, and interleukins 4, 6, and 8 [83–85]. These cytokines in turn induce inflammation, recruitment and activation of fibroblasts and myofibroblasts, leading to excess collagen production and reduced matrix metalloproteinase (MMP) activity [86].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


