Fig. 3.1
Overview of clinical trial setup in the academic setting. Diagram showing the workflow for initiation, submissions and approvals needed for a clinical trial
After IRB review, there may be queries that must be addressed prior to approval. In some cases, the protocol may need to be modified based on IRB recommendations. One of the critical components of the protocol is the informed consent form, which should be written in lay language, and clearly state the purpose of the research study. In addition, the informed consent form should include study-related procedures, risks and benefits of study participation, any costs to the participant and the human subjects’ rights (including the right to withdraw from the study at any time). The informed consent form may need to be amended as new information on the risks or benefits of a study drug become available. Adequate time to read the consent form and ask questions should be given. The informed consent form is signed by the potential study participant (or legally authorized representative) and a trained research staff member. If the potential participant is under 18 years of age, a parent or guardian signs the informed consent form, and the minor signs an assent form, if they are able.
3.5 Investigational New Drug Application
The United States Food and Drug Administration website (http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/InvestigationalNewDrugINDApplication/default.htm) provides a guide of whether human research studies require an investigational new drug application (IND). A study may be exempt from IND application if it is lawfully marketed in the USA, is not intended to support significant change in labeling or advertising of a drug and is not administered in a different route than the currently approved one (such as topical to oral administration) [4].
3.5.1 Drug Versus Device Trials
The FDA considers a drug a product whose primary intended use is “achieved through chemical action” whereas a device is a product used as an instrument or apparatus for prevention, diagnosis, treatment. For dermatology, these include dressings, adhesives for superficial skin lacerations, negative pressure wound therapy, and suture material. The safety and effectiveness of a device can be tested in a clinical trial. If the device is a significant risk to study participants, an “investigational device exemption” (IDE) can be submitted to the FDA. The device is classified by level of risk. IDE pre-submission meetings with the FDA to identify the type of scientific evidence needed for approval can be arranged. This can include clinical trial design and scope. Prior to commencement of a clinical trial, device studies require IRB approval as well as IDE exemption approval [5]. Subsequently, devices that are novel or have significant risk can be submitted to the FDA for a Premarketing Application (PMA) through clinical trials to approve a device for an intended use.
3.6 Budget and Contract Development
Once a protocol is finalized, a budget for the study can be estimated. At academic institutions this involves close collaboration with a budget development office where pre-determined prices for procedures and supplies can be obtained. Budget considerations include the amount of time allocated for a particular study, number and type of staff needed (e.g., biostatistician, research coordinator), cost of study agents, cost of clinic rooms, cost of supplies such as skin biopsy kits, cost of monitoring the study, and IRB costs.
For studies that involve industry partners, contract development with the legal team representing both the industry partner and the academic institution must be in place prior to study start. Elements of the contract include budget, indemnification (responsibility for any lawsuits or claims as a result of the clinical trial), and study duration.
3.7 Regulatory Issues
Regulatory issues pertain to compliance with government requirements for clinical trials. Documentation of measures taken to comply with regulations are compiled in a “regulatory binder.” The regulatory binder serves as a repository for study documents and includes the following elements: FDA 1572 (Statement of the Investigator), IRB approval letters and correspondence, signature and delegation log of all study staff, screening and enrollment lists, all versions of protocol and investigator brochures, all correspondence with industry sponsor and FDA, curriculum vitae of all study staff and their required training, list of protocol deviations and associated memoranda detailing the deviations, reportable serious adverse events (SAE), certificates of accreditation from clinical laboratories, specimen logs, copy of normal ranges of laboratory values. A helpful way to organize these sections would be to place the commonly used items towards the beginning. The regulatory binder is continuously updated during the course of the study [1–3].
3.8 Study Start-Up
The contract agreement needs to be finalized and routes for billing study costs established. Institutional account numbers need to be set up for the study. All required regulatory documents should be in place, including an IRB approval letter and consent forms that have not expired. All source documentation should be created with checklists for all procedures associated with study visits (Fig. 3.2). Case report forms to capture the critical data points should be in place. Study drug availability and dispensing should be confirmed from the investigational pharmacy prior to study start.
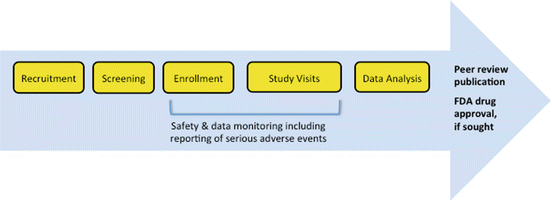
Fig. 3.2
Schematic of clinical trial conduct. Overview of clinical trial conduct from patient recruitment to publication and/or new drug development
All study staff need to be familiar with the protocol and have a chance to ask questions if anything is unclear. Industry-sponsored studies usually have a site initiation visit or meeting to review the protocol and answer questions.
Clinical trials that are interventional in nature and involve drugs or devices must be listed on the publicly available national website (www.clinicaltrials.gov). This is a searchable database for health care providers, patients and their caregivers to find studies that might be appropriate for a particular dermatologic condition. The studies are regularly updated, including information on whether active recruitment or enrollment is occurring, the inclusion and exclusion criteria, locations of centers where the study is being conducted, and results of the study when they become available.
3.9 Recruitment
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


