, Teresa S. Wright2, Crystal Y. Pourciau3 and Bruce R. Smoller4
(1)
Department of Pathology & Immunology, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(2)
Departments of Dermatology and Pediatrics, University of Tennessee Health Science Center, Memphis, TN, USA
(3)
Departments of Dermatology and Pediatrics, Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas, USA
(4)
Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
16.1 Calcinosis Cutis
16.1.1 Clinical Features
Calcinosis cutis is a rare phenomenon characterized by calcium salt deposition in the skin [1]. Skin lesions present as indurated subcutaneous plaques and nodules with or without associated erythema and tenderness. They may ulcerate and extrude creamy or chalky material, and can cause pressure erosions of underlying bones (Fig. 16.1). Calcinosis cutis is subdivided into four groups based on the etiology of deposition: dystrophic, metastatic, iatrogenic, and idiopathic.
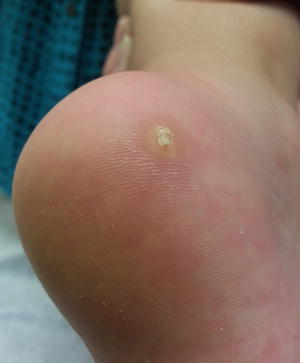
Fig. 16.1
Calcinosis cutis (heel stick calcinosis) is a firm, skin-colored nodule extruding chalky white material on the heel of a premature infant (photo courtesy of Audrey Chan, MD, Baylor College of Medicine, Houston, TX)
Dystrophic calcinosis cutis is the most common variant, occuring in areas of preexisting skin disease in the setting of normal phosphocalcium metabolism [1]. For example, dystrophic calcinosis cutis has been described in autoimmune connective tissue diseases, such as dermatomyositis and systemic sclerosis [2]. An alternative type of dystrophic calcinosis cutis is heel stick calcinosis or calcified nodules on the heels. This entity is seen in low birthweight infants after frequent heel pricks during the neonatal period. The skin changes tend to appear at 4–12 months of life with spontaneous resolution within 18 to 30 months of occurrence.
Metastatic calcification occurs in previously normal tissue following systemic disturbance with hyperphosphatemia and/or hypercalcemia [1]. Metastatic calcinosis is seen in individuals with hypervitaminosis D, sarcoidosis, osteolytic bone disease, and chronic renal failure.
Iatrogenic calcinosis has been reported following parenteral administration of solutions containing calcium or phosphate, as well as after electrophysiologic studies with calcium-impregnated electrode pastes [1].
Idiopathic calcinosis occurs without preceding evidence of tissue or metabolic disease. The most common variants of this subtype in children are milia-like calcinosis cutis of Down syndrome, idiopathic calcinosis of the scrotum, and subepidermal calcified nodules [1, 3]. Subepidermal calcified nodules can present during childhood or at birth, most commonly on the face or ears. Lesions may be persistent, requiring surgical excision .
16.1.2 Histology
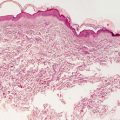
In calcinosis cutis, deposits of blue material that may appear spherular or fractured are surrounded by granulomatous inflammation (Fig. 16.2). In cases with dystrophic calcification, areas of follicular or eccrine disruption with keratinaceous debris may be seen adjacent to foci of calcium deposition. This is a frequent finding in pilomatricomas that occur in children and can also be seen in syringomas [7, 8]. Milia formation can also give rise to small calcium deposits in the dermis [9]. In many cases, the overlying epidermis becomes acanthotic and hyperkeratotic, suggesting lichen simplex chronicus-like changes. Ulceration with transepidermal elimination may be present [10]. Particularly in children, widespread dermal calcium deposits may be seen as a complication of dermatomyositis [5, 11–14]. In some cases, the etiology for the calcium deposition is not readily apparent [15]. Occasionally, calcium deposits are seen within infectious granulomatous processes [16].
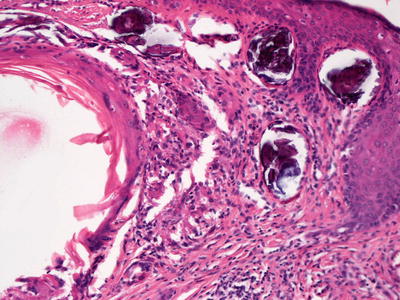
Fig. 16.2
Calcinosis cutis with calcium deposition within the dermis
16.1.3 Pathogenesis
Calcinosis cutis is caused by the deposition of insoluble calcium salts in the skin and subcutaneous tissue due to abnormalities in serum calcium and phosphorus levels [17]. Dystrophic calcification is caused by tissue damage that induces the release of phosphate-binding proteins by necrotic cells, resulting in calcified deposits that contain hydroxyapatite and amorphous calcium phosphate [18]. Proinflammatory cytokines such as interleukin-6 (IL-6) , IL-1, and tumor necrosis factor play an important role in this process [17, 19, 20]. Metastatic calcification is characterized by abnormally high levels of serum calcium and/or phosphorus, resulting in the precipitation of calcium salts in normal tissue with no structural damage. The levels of serum phosphorus affect the number and size of calcium deposits in tissues. The most frequent cause of metastatic calcification is chronic kidney failure with hyperphosphatemia. Other causes include hyperparathyroidism, sarcoidosis, and malignancies [21, 22].
16.2 Calciphylaxis
16.2.1 Clinical Features
Calciphylaxis affects 1–4 % of patients with end-stage renal disease [23]. Clinical associations include hyperparathyroidism, elevated calcium-phosphate product, hemodialysis, hypercoagulability, and renal transplantation. It is rare in children, with fewer than 10 reported cases, all of which occurred in boys [23, 24]. This differs from the demographics seen in adults, in which there is a striking female predilection.
Initial presenting clinical features include violaceous subcutaneous nodules or plaques, as well as reticulate erythema, which progress to cutaneous ulceration (Fig. 16.3). Most patients have severe recalcitrant pain and hyperesthesia associated with the skin changes. In children, lesions are seen with equal occurrence in the arms and legs with increased incidence of distal involvement. In contrast, in adults, skin lesions are most often localized to the proximal trunk and lower extremities [23].
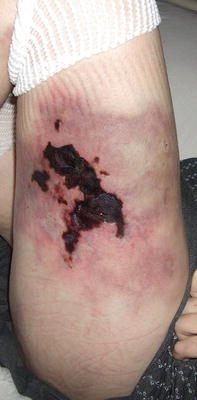
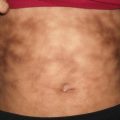
Fig. 16.3
Calciphylaxis presents as stellate ulceration with an eschar overlying an ill-defined, purpuric and indurated plaque on the thigh of a girl with end-stage renal disease
The clinical course of calciphylaxis is often marked by poor treatment response and non-healing ulcerations, predisposing individuals to gangrene. Mortality rates range from 60–80 % with most patients succumbing to sepsis within months of diagnosis [23]. Acral calcification portends a worse prognosis in children .
16.2.2 Histology
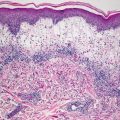
Histologic changes can be quite focal and subtle, or can be extensive [25]. In early lesions, calcium deposits may be identified solely within the walls of blood vessels. Vessels located in the deep dermis or subcutaneous fat are commonly affected (Fig. 16.4). There is little or no associated inflammatory infiltrate associated with the calcium deposits. In more extensive cases, calcium deposits also appear within the fat, unrelated to vascular deposits (Fig. 16.5) [26]. Peri-eccrine calcium deposition is a highly specific finding [27]. In severe cases, ulceration is present secondary to vascular occlusion and tissue ischemia. In these cases, a brisk and diffuse inflammatory process is observed. Interestingly, in one series, there was little evidence of internal organ calcium deposition at postmortem examination [28]. The histologic differential diagnosis includes atherosclerotic changes with secondary calcification, but this is not a frequent consideration in children.
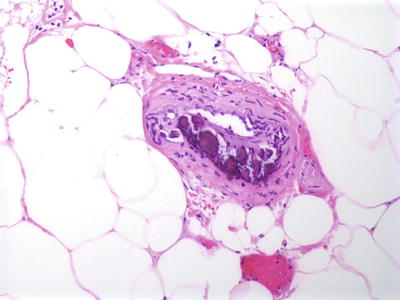
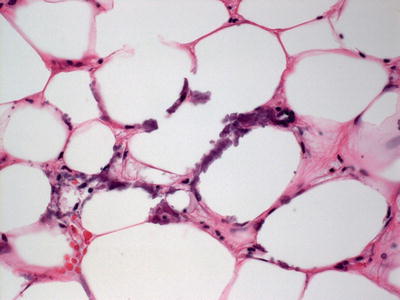
Fig. 16.4
Calciphylaxis is characterized by calcium deposits within the wall of blood vessels located in the deep dermis and subcutis
Fig. 16.5
Calcium deposits are seen in subcutaneous adipose tissue away from blood vessels in some cases of calciphylaxis
16.2.3 Pathogenesis
Calciphylaxis is a manifestation of severely dysregulated calcium–phosphorus metabolism, usually occurring in patients on dialysis for kidney failure [29]. There is a high prevalence of mineral bone abnormalities in dialysis patients. Kidney failure is known to affect calcium and phosphate status that can lead to calciphylaxis. Parathyroid hormone abnormalities may also be a cofactor in the pathogenesis of the disease since chronic hyperparathyroidism causes high bone turnover, hypophosphatemia, hypercalcemia, and vascular calcium deposition [29, 30].
Thrombotic vascular occlusion leads to tissue ischemia and necrosis in calciphylaxis [29]. There are reduced blood flow, vascular endothelial injury, and hypercoagulation state in this disorder. Calcium deposition and subintimal fibrosis involving small arteries lead to luminal narrowing and blood stasis. Inflammatory mediators, such as tumor necrosis factor-α and interleukin (IL)-1 and IL-6, are increased in chronic dialysis. These factors mediate endothelial cell dysfunction and promote a pro-coagulant state by vascular release of tissue factor, reduced protein C and protein S levels, and decreased thrombomodulin and heparin-like molecules [31, 32]. In addition to a localized pro-coagulant state, some patients with calciphylaxis also have systemic hypercoagulation [33, 34]. This may increase thrombus formation in calcified blood vessels.
A major molecular factor in calciphylaxis is nuclear factor-κB (NF-κB) and the receptor activator of NF-kB (RANK) and its ligand (RANKL) [35]. Agents, such as hormones, medications, and cytokines that affect NF-κB function can result in vascular calcification. Inhibitors of the RANK-NF-κB cascade, such as bisphosphonates and anti-RANKL antibodies, have the potential to prevent vascular calcification in calciphylaxis [35, 36].
16.3 Osteoma Cutis
16.3.1 Clinical Features
Osteoma cutis is a rare condition characterized by bone formation within the skin [37]. The condition can be subdivided into primary and secondary forms depending on the absence or presence of a preceding neoplastic process or cutaneous inflammation [37]. Primary disease is further subcategorized into progressive and limited forms. Osteoma cutis is characterized by subcutaneous rock-hard plaques or nodules with distribution dependent on the subtype of disease (Fig. 16.6). Lesions tend to be persistent, although a number of medical and surgical management options exist [37, 38].
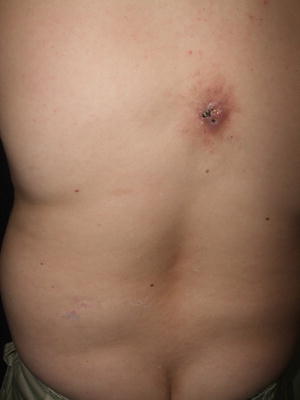
Fig. 16.6
Osteoma cutis presents as subcutaneous rock hard plaques and nodules on the back of a teenage boy with Albright hereditary osteodystrophy
16.3.2 Histology
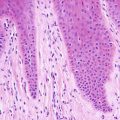
Osteoma cutis represents small bone deposits located in the dermis. In many cases, well-formed bone, even containing marrow, is present in the dermis without eliciting an inflammatory response (Figs. 16.7 and 16.8). In other cases, a granulomatous response can be seen. Osteoma cutis is an end stage of the dystrophic calcification process [39–41]. This is especially well described in some patients as a consequence of acne vulgaris. In such cases, the usual histologic changes of acne vulgaris may still be present in the biopsy [42].
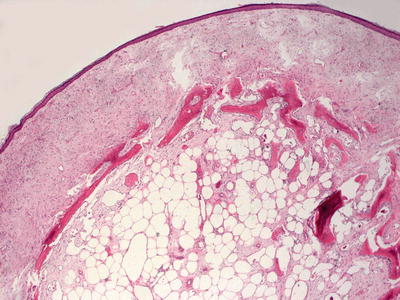
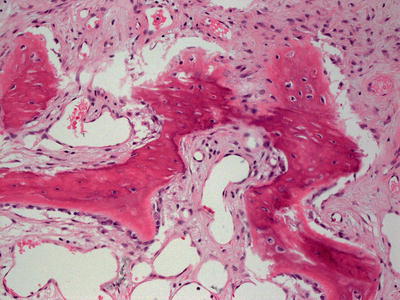
Fig. 16.7
Irregular osteoid formation is present in the dermis in osteoma cutis
Fig. 16.8
Osteoma cutis demonstrates osteoid formation within the dermis. In well-developed cases, apparent marrow can be seen within the osteoid deposition
Primary dermal osteoid formation occurs as part of several syndromes [39, 43–46]. One of these involves a plate-like deposition of bone within the dermis, and epidermolytic hyperkeratosis may be seen in the overlying epidermis [41, 47, 48]. Another syndrome involves café-au-lait spots and woolly hair anomalies in association with osteoid deposits in the dermis [49]. In rare cases, patients with metabolic disorder or endocrinopathies may develop osteoid deposits in the dermis [50].
16.3.3 Pathogenesis
Osteoma cutis is caused by abnormal deposition of bone in the dermis. The pathogenesis of osteoma cutis is unknown, but it has been postulated to be associated with the metaplasia of fibroblasts into osteoblasts and subsequent bone production [51]. In situ hybridization studies have shown that dermal fibroblasts can differentiate into osteoblasts, and produce type 1 collagen and osteonectin characteristic of bone-forming cells [52]. Disorders of ossification in the skin may be linked to Albright hereditary osteodystrophy, progressive osseous heteroplasia, and congenital plate-like osteoma as well as disorders with non-syndromic mutations in GNAS gene [45, 53, 54]. GNAS is the α subunit of the G protein of adenylyl cyclase (Gsα), which promotes the differentiation of mesenchymal stem cells into osteoblasts. Other common causes of osteoma cutis include infection, surgical procedures, granulomatous diseases, tumors, and trauma [45, 53–55].
16.4 Erythropoietic Protoporphyria
16.4.1 Clinical Features
Erythropoietic protoporphyria is the most common type of porphyria seen in children [56]. This inherited porphyria is a result of partial deficiency in ferrochelatase with subsequent accumulation of the photoreactive metabolite protoporphyrin IX in erythrocytes, plasma, liver, and skin.
Symptoms typically present in infancy or early childhood with photodistributed skin pain, burning or itching immediately upon sun exposure [56]. Mild edema and erythematous to purpuric patches can be seen on the hands. In chronic disease, waxy indurated plaques and scarring can develop on the dorsum of the hands and the central face, specifically along the nasal bridge. Twenty-five percent of patients have involvement of the hepatobiliary system secondary to accumulation of protoporphyrin IX in the liver [56]. Ten to twenty percent of patients have liver dysfunction. Patients are also at risk for cholelithiasis with 2 % of affected individuals developing acute fulminant cholestatic liver failure.
16.4.2 Histology
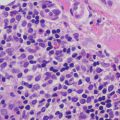
There is a cell-poor subepidermal blister with prominent festooning of the papillary dermal tips in erythropoietic protoporphyria (Figs. 16.9 and 16.10). Within the stratum corneum, eosinophilic bodies that have been called “caterpillar bodies ” may be observed when intact epidermis is available for examination. These elongated and segmented figures comprise laminin and type IV collagen [57]. Acute lesions demonstrate basal vacuolization in the epidermis with intercellular edema. Cytolysis of endothelial cells is also present. As the lesions become more chronic, PAS-positive hyaline material is deposited around dermal blood vessels (Fig. 16.11). The capillary basement membranes are also thickened [58, 59]. Late stage lesions show extensive reduplication of vascular basement membranes on electron microscopic examination [58]. Direct immunofluorescence examination shows linear IgG, IgA, IgM, and C3 concentrically around the dermal blood vessels [59].
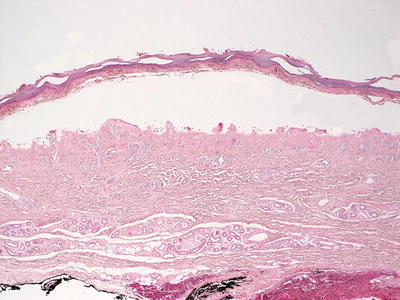
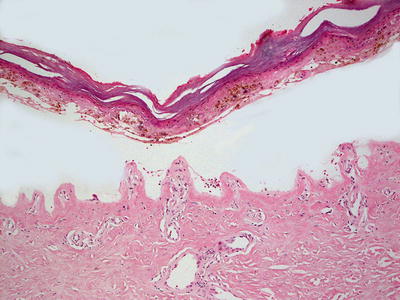
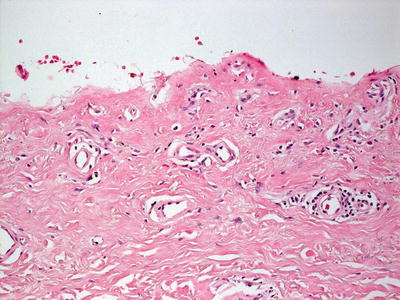
Fig. 16.9
Erythropoietic protoporphyria demonstrates a noninflammatory subepidermal blister
Fig. 16.10
Prominent festooning of the papillary dermal tips into the blister cavity is a frequent finding in erythropoietic protoporphyria
Fig. 16.11
Erythropoietic protoporphyria demonstrates dilation of papillary dermal blood vessels that are often lined by a rim of eosinophilic material
Lipoid proteinosis can be distinguished from erythropoietic protoporphyria by a more diffuse deposition of hyaline material than that seen in porphyria [61]. Deposition of hyaline material is a frequent finding in lipoid proteinosis, but it is not seen in erythropoietic protoporphyria. Clinical presentation also makes the distinction relatively simple. Porphyria cutanea tarda demonstrates similar hyaline deposits around dermal blood vessels, but it does not have the marked thickening in capillary walls due to basement membrane reduplication as seen in erythropoietic porphyrias .
16.4.3 Pathogenesis
Erythropoietic porphyrias are characterized by elevations of porphyrins in the bone marrow and red blood cells with skin photosensitivity that develops in infancy or early childhood [62, 63]. The mechanism of cutaneous photosensitivity in erythropoietic porphyrias is due to the accumulation of excess protoporphyrin in the skin and cutaneous blood vessels, and photoactivation of protoporphyrin by sunlight.
Congenital erythropoietic porphyria (CEP) is an autosomal recessive disorder associated with hemolytic anemia and severe skin photosensitivity. CEP results from deficiency in the activity of uroporphyrinogen III synthase due to mutations in the GATA1 and CP2 transcriptional binding elements in the promoter region of uroporphyrinogen III synthase gene. Deficient uroporphyrinogen III synthase leads to the accumulation of uroporphyrin I and coproporphyrin I isomers [62, 64, 65]. Patients with CEP that have uroporphyrinogen III synthase mutations plus gain-of-function mutations in the 5-aminolevulinate synthase 2 (ALAS2) gene, which encodes for a rate-limiting enzyme of erythroid heme synthesis, have a more severe disease phenotype than patients without concomitant ALAS2 mutations [66]. An X-linked variant of CEP has been identified that is caused by mutation in GATA1 gene, which encodes a transcription factor that regulates the expression of globin and heme biosynthetic genes [67].
Erythropoietic protoporphyria is one of the most common erythropoietic porphyrias in children [62]. It is an autosomal recessive disorder caused by loss of function mutations in the ferrochelatase (FECH) gene. FECH is an enzyme in the heme biosynthetic pathway. Defects in FECH lead to increased levels of protoporphyrins in red blood cells. The protoporphyrins are not complexed with zinc, and are free to bind to hemoglobin. There is an X-linked form of erythropoietic protoporphyria that is due to activating mutations in the ALAS2 gene with increased ALAS2 activity and accumulation of protoporphyrins. Mutational analyses for FECH and ALAS2 genes establish the genetic diagnosis for the disease [62].
16.5 Tattoo
16.5.1 Clinical Features
Approximately 7–20 million people in the United States are estimated to have at least one tattoo with a prevalence rate of 10–26 % in men and 10–22 % in women [68]. Prevalence is relatively similar across different ethnic groups [69]. The appearance of tattoos is varied and characterized by the permanent inking of colors in various patterns on the skin. They can be removed or lightened with lasers. However, risk of removal includes secondary dyschromia, especially in more darkly pigmented skin types. Graphite tattoos generally occur through traumatic implantation. In these situations, small areas of dermal pigmentation may be present (Fig. 16.12).
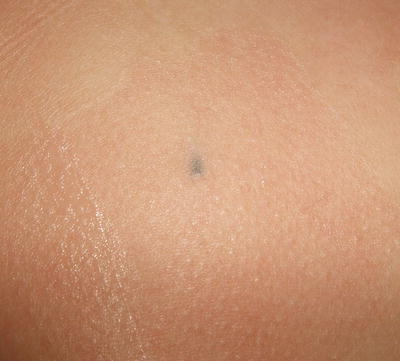
Fig. 16.12
Graphite tattoo shows a bluish gray macule on the chest following puncture wound with a lead pencil
16.5.2 Histology
Histologic findings in a tattoo include the observation of various pigments in the dermis (Figs. 16.13 and 16.14). The pigments tend to aggregate around blood vessels, and can be present within macrophages and in the interstitial space. The pigment can be relatively superficial or extend much deeper into the dermis. In some patients, there is little immunologic reaction to the pigment. However, tattoos are often associated with immune reactions.
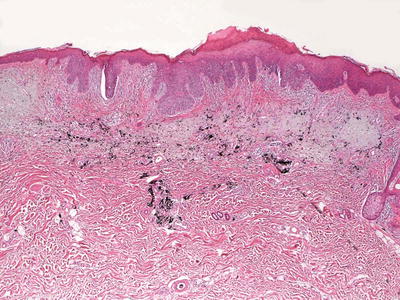
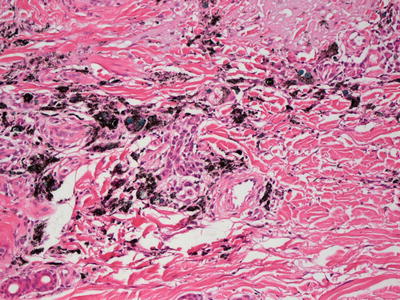
Fig. 16.13
Dermal pigment is present in a tattoo , often centered on dermal blood vessels. The pigment may be seen in the superficial and deep dermis
Fig. 16.14
It is often possible to identify various colors in a tattoo
One of the most common reactions to a tattoo is the presence of overlying pseudoepitheliomatous hyperplasia [70–74]. This reaction pattern can be so florid as to raise the possibility of a squamous cell carcinoma [72]. A rare reaction pattern is a chronic fibrosing vasculitis-type of pattern that is localized entirely within the tattoo, and specifically to areas with red pigment [73]. An intense lichenoid inflammatory reaction has been described in response to red pigment within a tattoo [75]. Acute contact dermatitis, lupus-like inflammatory reactions, and foreign body granulomas are also described as immune responses to tattoos [76]. Necrobiotic granuloma-like reactions to tattoos are also seen, but they are far less common that foreign body type granulomas [77]. A florid cutaneous lymphoid hyperplasia resembling a lymphoma occurs as a reaction to tattoos with the majority of lymphocytes in these hyperplastic responses are T cells [78, 79]. Interestingly, this reaction is also more commonly associated with red pigment. A rare tattoo reaction resembling cutaneous morphea has also been reported [80].
16.6 Papular Mucinosis
16.6.1 Clinical Features
The exact incidence of papular mucinosis is unknown with some authors suggesting that occurrence is underreported [81]. Papular mucinosis is characterized by small indurated, waxy papules, usually localized to the trunk and proximal extremities [82]. Lesions may coalesce to form nodules and plaques.
Papular mucinosis can be subcategorized based on the clinical appearance. The variants most commonly seen in children are self-healing juvenile papular mucinosis and papular mucinosis of infancy. Self-healing juvenile papular mucinosis is characterized by the rapid onset of linearly arranged papules that coalesce into plaques on the face, neck, scalp, abdomen, and thighs. The reported age of onset ranges between 13 months and 15 years of age [82, 83]. Systemic complaints of fever, arthralgias, and weakness may occur with onset of skin lesions, although there is no association with an underlying systemic disease. In contrast, papular mucinosis of infancy is described as firm, shiny papules at the upper arms and trunk, particularly along the elbows. The condition tends to be persistent, and is not associated with underlying paraproteinemia or other systemic abnormalities [82, 84]. As most cases of papular mucinosis are not associated with underlying systemic disease, treatment is not required. The self-healing variant often resolves within weeks to months with similar reports of eventual spontaneous resolution noted in the infantile form as well [82].
16.6.2 Histology
The precise nature of papular mucinosis of infancy is still very much in question with some investigators favoring a discrete entity, and others considering this to be an early presentation of lichen myxedematosus [81, 84–86]. The histologic findings include an accumulation of mucin within the superficial portion of the dermis accompanied by an increased number of dermal fibroblasts. However, in some cases, fibroblasts are not significantly increased. The epidermis is either unremarkable or slightly flattened due to the accumulation of dermal mucin. There is a mild lymphohistiocytic inflammatory infiltrate in some cases. Older lesions demonstrate thickening of collagen bundles.
Self–healing juvenile cutaneous mucinosis is a rare disorder that is characterized by dermal mucin deposition, usually in the absence of increase in dermal fibroblasts (Fig. 16.15). Some cases show a slight increase in fibroblasts and mast cells [87]. Mucin deposition predominates in the superficial portion of the reticular dermis [88, 89] (Fig. 16.16). The accumulation of mucin is relatively well circumscribed, and can be better visualized on colloidal iron stains [90].
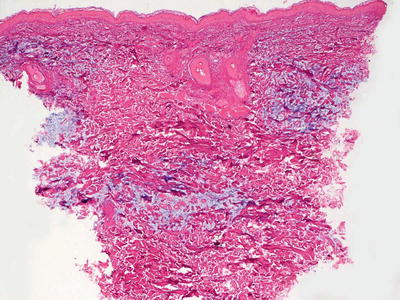
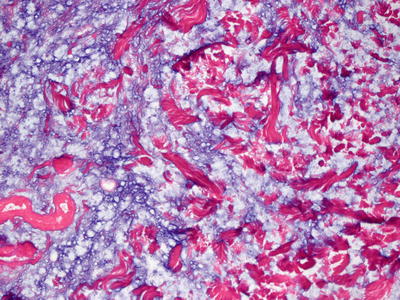
Fig. 16.15
Routine histologic sections demonstrate increased mucin diffusely throughout the dermis in self-healing juvenile cutaneous mucinosis
Fig. 16.16
Dermal mucin is readily apparent with routine staining in most cases of self-healing juvenile cutaneous mucinosis
A rare variant has been described in which mucin deposition is deeper and more extensive, extending into the subcutis. The histologic appearance is quite similar to nodular fasciitis or proliferative fasciitis with relatively inconspicuous mucin deposits [83, 91]. Myxoid stroma and gangliocyte-like giant cells may be present in this rare variant [83]. A single case with apparent disease progression of the self-healing condition, as opposed to spontaneous remission, exists in the literature [92].
The differential diagnosis includes other cutaneous mucinoses, such as scleromyxedema and lichen myxedematosus. While these entities generally display a more excessive increase in dermal fibroblasts than that seen in self-healing juvenile cutaneous mucinosis, resolution of the dilemma usually resides in a thorough clinical work-up to exclude any systemic involvement, and close clinical follow-up to monitor the course of the disease .
16.7 Digital Mucous Cyst
16.7.1 Clinical Features
Digital mucous cysts are benign ganglion cysts that are rare in children, and most commonly occur in adults between 40 and 70 years of age [93]. Females are affected twice as often as males [94]. Lesions typically appear as an isolated skin-colored to slightly erythematous nodule on the dorsum of the digit along the distal interphalangeal joint or proximal nail fold. The hands are affected more often than the feet [93]. Clustered or multiple lesions have been described. As most cysts are asymptomatic, no treatment is required but surgical management can be offered.
16.7.2 Histology
A digital mucous cyst consists of a collection of alcian blue or colloidal iron-positive ground substance present in the superficial dermis (Figs. 16.17 and 16.18). Despite the name, there is no cyst lining present. Surrounding the pseudocyst is a fibrous and myxoid stroma with increased numbers of fibroblasts [94, 95]. Alcian blue or colloidal iron stain demonstrates pools of mucin within the dermis (Fig. 16.19). The epidermis is usually unremarkable. The histologic findings in a digital mucous cyst are quite similar to those seen in a ganglion cyst. The anatomic site determines the diagnosis.
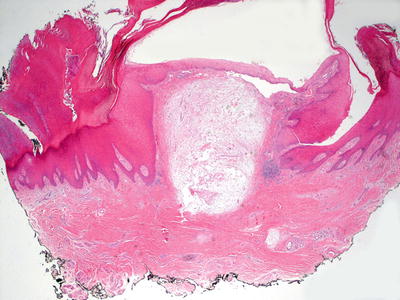
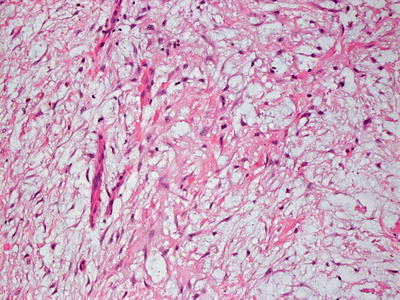
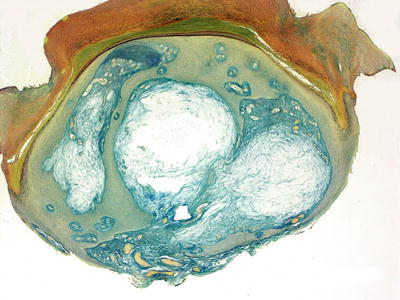
Fig. 16.17
Digital mucous cyst is a pseudocyst that demonstrates a collection of mucin within the papillary and reticular dermis of acral skin
Fig. 16.18
Increased dermal mucin is readily apparent with routine H&E staining of digital mucous cyst
Fig. 16.19
Alcian blue stains highlight the loculated collections of dermal mucin in digital mucous cyst
16.7.3 Pathogenesis
Digital mucous cyst does not have a true epithelial lining, so it is more accurately classified as a pseudocyst. The superficial (mucinous) type is thought to arise from metaplasia of dermal fibroblasts with excess production of hyaluronic acid and mucin [96, 97]. The deep (ganglion) type is thought to result from degenerative changes in the fibrous capsule or synovial tissue of the joint with excess production and accumulation of hyaluronic acid through a communication channel or peduncle to form a digital mucous cyst [98, 99]. Radiographic and surgical evidence has shown communication between the pseudocyst and adjacent joint space [100, 101]. There is evidence of the association of digital mucous cyst with osteoarthritis. It has been suggested that the development of osteophytes, and an increase in synovial fluid in osteoarthritis contribute to the cyst formation [97, 102, 103].
16.8 Scleromyxedema/Lichen Myxedematosus
16.8.1 Clinical Features
Scleromyxedema is a rare disorder usually of middle-aged adults with no gender predilection [81]. It is typically associated with an underlying systemic disorder, most commonly paraproteinemia, which is seen in approximately 83 % of cases [81]. Scleromyxedema typically presents as widespread coalescent shiny papules, often in linear arrangement distributed symmetrically on the hands, forearms, face, neck, upper trunk, and thighs. Indurated longitudinal furrowing may be seen along the glabella. With progression of disease, erythematous indurated plaques may develop with concomitant skin-stiffening and decreased mobility. The mucous membranes and scalp are spared with absence of telangiectases and calcinosis.
Treatment of the condition is difficult, but cutaneous lesions may resolve with successful management of the underlying disorder. Approximately 10 % of patients with an associated monoclonal gammopathy eventually develop multiple myeloma [81]. Additional systemic sequelae include other hematologic malignancies, proximal muscle weakness, central nervous system disturbances, joint disease, Raynaud’s phenomenon , pulmonary disease, dysphagia, laryngeal involvement, scleroderma-like renal disease, and ocular changes .
16.8.2 Histology
Scleromyxedema and lichen myxedematosus have identical histologic findings, and are differentiated based upon the clinical presentation and associated systemic symptoms [81, 86]. The superficial dermis is distended with mucin (acid mucopolysaccharides) that is apparent on routine histologic sections, and can be better visualized on colloidal iron or alcian blue stains [104] (Figs. 16.20 and 16.21). There is an increase in dermal fibroblasts. In more established lesions, thickening of dermal collagen further increases the thickness of the dermis. A proliferation of eccrine ducts can be seen in some cases [105]. Degradation of collagen has been demonstrated by ultrastructural examination [106]. A rare variant of scleromyxedema with foci histologically resembling the interstitial type of granuloma annulare has been described [107, 108].
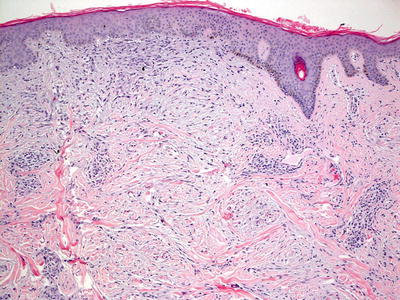
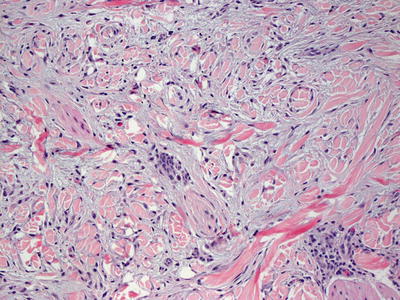
Fig. 16.20
Increased numbers of fibroblasts are present in the papillary and superficial reticular dermis in scleromyxedema
Fig. 16.21
Dermal mucin is seen interspersed between collagen bundles within the superficial dermis in scleromyxedema
Involvement of the fibrous septa is quite unusual in scleromyxedema, and in most cases, deep extension can be used to favor a diagnosis of nephrogenic systemic fibrosis over lichen myxedematosus/scleromyxedema. It has been proposed that the presence of iron within dermal fibroblasts as seen in nephrogenic systemic fibrosis might aid in making this histologic distinction [109]. Careful attention to clinical history is the easiest way to distinguish between these entities that have great histologic overlap [110]. Scleroderma is differentiated based upon the lack of dermal mucin, and scleredema lacks a proliferation of fibroblasts within the dermis. Furthermore, mucin deposition in scleredema is more subtle and deeper within the reticular dermis. Pretibial myxedema can be differentiated from scleromyxedema based upon the clinical presentation, and the lack of significant fibroblast proliferation to accompany the dermal mucin deposition .
16.8.3 Pathogenesis
Scleromyxedema is a generalized form of dermal mucin deposition, and it is often associated with monoclonal gammopathy, particularly of immunoglobulin G [111, 112]. Serum from patients with scleromyxedema has been shown to enhance the proliferation of skin fibroblasts in vitro, but immunoglobulins purified from the serum do not affect fibroblast growth [113, 114]. Therefore, other serum factor(s), and not immunoglobulins, may be responsible for the enhanced fibroblast proliferation, and the subsequent induction of mucin deposition in the skin. Intrinsic dysregulation of skin fibroblasts in scleromyxedema may be another possible mechanism of increased fibroblast proliferation .
16.9 Pretibial Myxedema
16.9.1 Clinical Features
Up to 4 % of patients with thyrotoxicosis of Graves’ disease , and 13 % of those with more advanced ophthalmopathy develop thyroid dermopathy and pretibial myxedema [115]. Pretibial myxedema is characterized by symmetric circumscribed, light-colored to brownish yellow, non-pitting, indurated plaques at the pretibial legs and feet [115]. Follicular ostia may become more prominent with a peau d’orange appearance of affected skin. Nineteen to twenty-five percent of patients with severe thyroid dermopathy will develop thyroid acropachy, presenting as clubbing of the digits. Eighteen percent of patients will develop a nodular variant with only 5 % developing the most severe subtype with coalescent nodular, fungating plaques (elephantiasis). Hyperhidrosis is a common feature, while pruritus and tenderness are infrequently seen.
The likelihood of disease remission is inversely correlated with the severity of disease at initial presentation [115]. Specific treatment of the skin changes may not be required as mild dermopathy may demonstrate partial or complete resolution over time .
16.9.2 Histology
Histologic findings in pretibial myxedema include the presence of increased dermal mucin located primarily in the superficial reticular dermis (Fig. 16.22). A grenz zone within the papillary dermis is reported to be associated with Graves’ disease, although it is usually absent in patients with pretibial myxedema in the absence of this disease [116]. Mucin extends deeper into the reticular dermis in patients with Graves’ disease. Only a minimal increase in dermal fibroblasts is seen in most cases (Fig. 16.23). A mild lymphohistiocytic inflammatory response may be present [117]. The overlying epidermis may range from normal to displaying verrucous epidermal hyperplasia with overlying hyperkeratosis. A rare variant of pretibial myxedema demonstrates a nodular aggregation of mucin within the dermis [118].
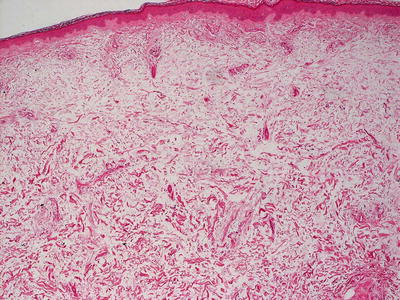
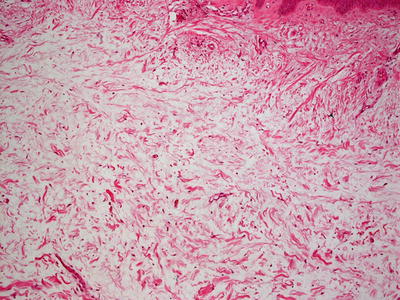
Fig. 16.22
Pretibial myxedema shows increased amount of mucin in the superficial portion of the dermis. Mucin may also be present in the deeper dermis in extensive cases
Fig. 16.23
Increased mucin is seen in the dermis in pretibial myxedema . There is no appreciable increase in dermal fibroblasts in this disorder
The histologic differential diagnosis includes other mucinoses. The mucin deposition in scleromyxedema and lichen myxedematosus is accompanied by a brisk fibroblastic infiltrate not seen in pretibial myxedema. Scleredema shows mucin in the deep reticular dermis, and the deposition is less apparent on routine H&E sections. Stasis dermatitis may also present with mucin in the superficial dermis, but this is an uncommon finding in children [116].
16.9.3 Pathogenesis
Pretibial myxedema is a skin manifestation of autoimmune Graves’ disease [119, 120]. Activation of fibroblasts with excessive production of glycosaminoglycans is the main pathologic process in pretibial myxedema. Local accumulation of glycosaminoglycans leads to compression of dermal lymphatics and results in non-pitting edema [115, 121]. The mechanism of fibroblast activation in pretibial myxedema involves thyroid stimulating hormone (TSH) receptor -stimulating antibodies, and TSH receptors present in the skin that initiate the immune process. There is evidence that thyroid-specific T lymphocytes accumulate in pretibial tissues in patients with Graves’ disease and pretibial myxedema [122]. Cytokines produced by T lymphocytes can stimulate dermal fibroblasts to produce glycosaminoglycans and hyaluronic acids [119, 123]. Another potential mechanism of fibroblast activation involves the insulin-like growth factor-1 receptor (IGF-1R). IGF-1R acts as an antigen to Graves’ disease immunoglobulins, resulting in the upregulation of T-cell cytokines and fibroblast activation [124, 125].
16.10 Nephrogenic Systemic Fibrosis
16.10.1 Clinical Features
Nephrogenic systemic fibrosis is a rare disorder seen in patients with renal dysfunction following exposure to gadolinium-based contrast media [126]. There is no gender or racial predilection. It primarily occurs in middle-aged adults, with only a few published cases in children [127].
Nephrogenic systemic fibrosis is characterized by a coalescence of brawny, indurated papules and plaques, most often along the trunk and extremities with sparing of the face [126]. Patients may complain of concomitant pruritus or pain, especially if contractures develop over involved joint spaces. The clinical course is usually progressive, although spontaneous resolution may occur following recovery of normal renal function [127]. The associated skin fibrosis and joint contractures cause significant debilitation, but ultimately, prognosis is determined by the degree of systemic involvement [126, 127]. Fibrosis of the heart, lungs, diaphragm, skeletal muscles, and kidneys contribute most significantly to the mortality and morbidity of disease .
16.10.2 Histology
Biopsy findings include a fibroblastic deposition in the mid-reticular dermis extending down the fibrous septae into the subcutis (Fig. 16.24). There is accompanying mucin deposition throughout the dermis and subcutis. Early lesions appear edematous with a hypercellular appearance due to increased fibroblasts, while later lesions appear less cellular and have more sclerotic and eosinophilic collagen with less obvious mucin (Figs. 16.25, 16.26 and 16.27). Collagen bundles are thickened in late-stage lesions with decreased interstitial spaces. Small multinucleated giant cells have been described in some cases [128].
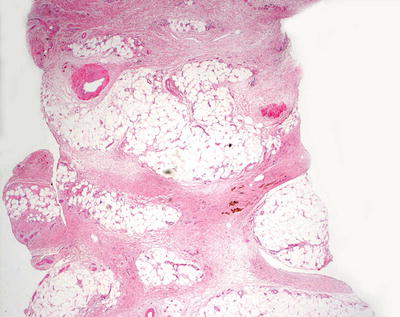
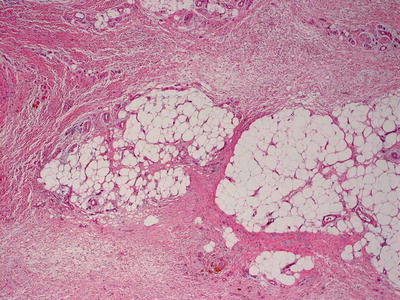
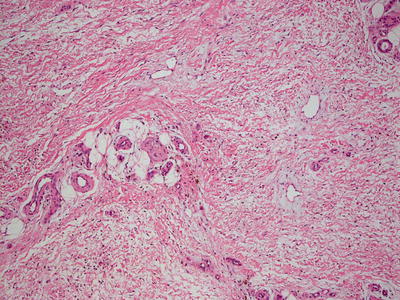
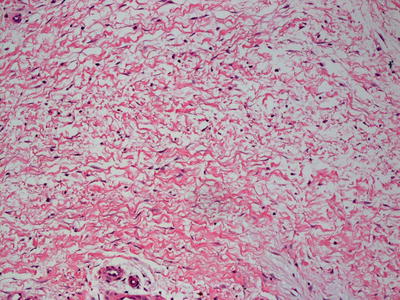
Fig. 16.24
Marked thickening of the dermal and subcutaneous collagen are seen in nephrogenic systemic fibrosis , giving rise to the appearance of a septal panniculitis in some cases
Fig. 16.25
Increased fibroblasts are seen within the thickened collagen in all levels of the dermis and subcutis in nephrogenic systemic fibrosis
Fig. 16.26
Increased ground substance consisting primarily of mucin is seen in nephrogenic systemic fibrosis
Fig. 16.27
The combination of increased cellularity and dermal mucin resulting in separation of collagen bundles in the deep dermis is characteristic of nephrogenic systemic fibrosis
The differential diagnosis of early lesions is scleromyxedema or lichen myxedematosus [110]. The clinical course and presentation are the easiest way to make the distinction, although nephrogenic systemic fibrosis involves the deep reticular dermis and subcutis, which are unusual findings in the other disorders. It has been suggested that the presence of hemosiderin in fibroblasts is more indicative of nephrogenic systemic fibrosis than scleromyxedema, and can be used as a discriminating feature [109]. Later lesions may bear some resemblance to scleroderma, but the increased number of fibroblasts is unusual in scleroderma. Scleredema involves the deep reticular dermis, but it is not associated with a hypercellular dermis .
16.10.3 Pathogenesis
Nephrogenic systemic fibrosis is an iatrogenic fibrosing disorder in patients with chronic kidney disease who have been exposed to gadolinium-based contrast agents during imaging procedures [129]. Instead of being cleared rapidly by the kidney, gadolinium–chelate complexes remain in the body longer in patients with impaired renal function. During this time, gadolinium can dissociate from its chelate and bind to tissue. Gadolinium deposition has been found in the skin and other tissues from patients with nephrogenic systemic fibrosis [130, 131]. In vitro studies have shown that gadolinium induces the production of pro-inflammatory and profibrotic cytokines by human macrophages and monocytes through Toll-like receptors (TLRs) 4 and 7 [132, 133]. Some key cytokines involved are interleukin (IL)-4, IL-6, IL-13, and interferon (IFN)-γ as well as nuclear factor-κB-dependent cytokines [132, 134]. These cytokines activate dermal fibroblasts to synthesize extracellular matrix proteins and induce fibrosis. Fibroblasts exposed to gadolinium become fully differentiated myofibroblasts that exhibit an activated phenotype and produce increased amounts of types I and III collagen, fibronectin, and hyaluronic acid [135]. A study has provided evidence that iron is important in the pathogenesis of nephrogenic systemic fibrosis in an animal model of the disease [136]. Iron chelation with deferiprone significantly improves gadolinium-induced nephrogenic systemic fibrosis-like skin changes in mice .
16.11 Tangier Disease
16.11.1 Clinical Features
Tangier disease is a rare autosomal recessive disorder with defects in lipid transport and accumulation of cholesterol esters in tissues, such as the tonsils. The skin usually appears normal, but can present with small firm papules.
16.11.2 Histology
Tangier disease is characterized by deposition of cholesterol esters within histiocytes surrounding blood vessels in the superficial dermis, leading to their having a foamy appearance [137, 138]. Vacuolization of Schwann cells within cutaneous nerves is apparent on H&E sections, and it is greatly accentuated in ultrathin sections or electron microscopic examination [139]. Similar vacuoles can be seen in melanocytic nevus cells and perineurial cells [140].
16.11.3 Pathogenesis
Tangier disease is a genetic disorder of deficiency of high-density lipoprotein (HDL) with less than 5 % of normal plasma HDL levels and increased incidence of early cardiovascular disease [141, 142]. The underlying molecular defect in Tangier disease is mutations in the ATP-binding cassette transporter-1 (ABCA1) [143–145]. Over 50 distinct mutations in ABCA1 have been identified in this disease. Most of the mutations lie in the extracellular domains and the nucleotide-binding domains of ABCA1 [146].
ABCA1 plays a key role in the formation of nascent HDL. ABCA1 is a transmembrane protein that functions as a cholesterol transporter, and is involved in the transport of lipids across the plasma membrane [147, 148]. ABCA1 is also a binding partner of apolipoprotein A-I (apoA-I) , which is the major protein constituent of HDL. Impaired cholesterol transfer to apoA-I is a major feature in cells from patients with Tangier disease [149, 150]. The interaction of ABCA1 with apoA-I promotes the efflux of phospholipids and cholesterols from the cell, and activates intracellular signal transduction, including the small G proteins Cdc42 and Rac1, and protein kinases PKA, JAK2, and p54JNK. These signaling pathways result in cholesterol transport and lipidation of apoA-I to form HDL [149, 151]. Since the interaction of ABCA1 with apoA-I as well as lipid efflux occurs in the plasma membrane, the inability of mutant ABCA1 to be correctly localized in the plasma membrane is a mechanism leading to defects in the formation of HDL .
16.12 Mucopolysaccharidoses
16.12.1 Clinical Features
The mucopolysaccharidoses (MPS) are an inherited group of metabolic disorders caused by defects in lysosomal hydrolases that are responsible for the degradation of glycosominoglycans [152]. Seven subtypes exist, with a worldwide incidence of approximately 1 in 25,000 individuals. The most common variant is Sanfilippo syndrome (MPS III) [153]. While most babies appear normal at birth, the condition is progressive with affected individuals demonstrating features of the disease in infancy. The most common clinical signs include coarse facies and brawny thickening of the skin with broad nose, macroglossia, short neck, and macrocephaly [153] (Fig. 16.28). Other common manifestations include hypertrichosis, organomegaly, skeletal abnormalities, impaired hearing, and varying levels of mental retardation. Cutaneous features of the disease may include widespread progressive congenital dermal melanocytosis as well as white to skin-colored papules and papulonodules along the back, particularly in types I and II MPS [154]. Disease prognosis varies depending upon the underlying subtype, but many affected individuals experience premature death secondary to complications from systemic involvement [152].
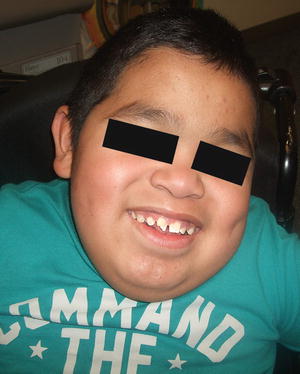
Fig. 16.28
A boy with Sanfilippo syndrome (mucopolysaccharidosis) demonstrates a number of the most common clinical features of mucopolysaccharidosis including macrocephaly, coarse facial features, broad nose, macroglossia, and a short neck
16.12.2 Histology
Ultrastructural studies are required to make a histologic diagnosis of MPS. The likelihood of diagnostic findings on ultrastructural analysis is quite high, and skin biopsy is an effective means for confirming such a diagnosis, while most routine studies fail to demonstrate significant abnormalities [155]. Occasionally, increased mucopolysaccharides can be seen as metachromatic “mucin deposits” within the dermis [156]. Dermal fibroblasts and macrophages demonstrate vacuolar inclusions that are either clear or have a fuzzy appearance [157–160]. These inclusions appear to be membrane bound. Similar observations have been made in keratinocytes, Schwann cells, and smooth muscle cells, as well as worm-like inclusions within mast cells [157]. Other cutaneous findings, such as angiokeratomas and Mongolians spots, are sometimes associated with mucopolysaccharidoses [161–163].
16.12.3 Pathogenesis
The mucopolysaccharidoses are genetic disorders of lysosomal storage diseases with impairment in glycosaminoglycans and mucopolysaccharides catabolism due to deficiency in activity of lysosomal enzymes responsible for the normal degradation of these molecules [152, 164, 165]. As a result, there is accumulation of glycosaminoglycans in the lysosomes in most cells as well as secretion of these polysaccharides in the blood, cerebral spinal fluid, and urine, leading to cellular and tissue damage and multi-organ failure [166–168].
There are seven types of MPS (I, II, III, IV, VI, VII, and IX) based upon the affected enzyme [152, 165]. Mucopolysaccharidosis type I (MPS I) is caused by a lack of a-L-iduronidase (IDUA) that is required for the breakdown of glycosaminoglycans, particularly heparan sulfate and dermatan sulfate. MPS II (also called Hunter syndrome ) is an X-linked genetic disorder caused by deficiency in iduronate-2-sulfatase (I2S), resulting in the accumulation of heparan sulfate and dermatan sulfate within the lysosome [164]. MPS III (also called Sanfilippo syndrome ) is caused by a lack of enzymes necessary for the degradation of heparan sulfate [169]. MPS IV (Morquio syndrome ) is caused by deficiency in N-acetylgalactose-amine-6-sulfate sulfatase (GALNS) and beta-galactosidase (GLB1), which are involved in the catabolism of keratan sulfate and chondroitin sulfate. MPS VI (Maroteaux–Lamy syndrome ) is caused by the lack of N-acetylgalactosamine 4-sulfatase (ASB) with accumulation of dermatan sulfate and chondroitin sulfate. MPS VII (Sly syndrome ) is caused by β-glucuronidase (GUSB) deficiency, and results in the deposition of chondroitin sulfate, heparan sulfate, and dermatan sulfate in the cell [170]. MPS IX (Natowicz syndrome) is caused by defects in hyaluronidase [165, 171].
16.13 Mucolipidoses
16.13.1 Clinical Features
The mucolipidoses are a group of metabolic disorders of variable phenotype caused by malfunction of a number of different enzymes responsible for appropriate lysosomal functioning [152]. Presentation and prognosis is dependent upon the affected enzyme. The most severe variant is mucolipidosis type II. Individuals with mucolipidosis type II present as neonates with severely malformed bones, microcephaly and dysmorphic facies, similar to that seen in Hurler syndrome [152]. Additional clinical features include severe neurologic impairment, respiratory difficulty, feeding problems, hernias, hepatosplenomegaly and cardiomyopathy, which portends a worse clinical course. Most patients die from cardiorespiratory complications by 10 years old .
16.13.2 Histology
Skin biopsies are generally helpful in establishing a diagnosis of mucolipidosis and other lysosomal storage disorders [155, 172–176]. Ultrastructural examination is most revealing, demonstrating lamellar and pseudomyelinitic structures in a variety of cell types [177]. Epithelial cells and endothelial cells can show membrane lamellar inclusions [178]. Vacuolated fibroblasts and macrophages may be seen in some cases [179]. Other cutaneous lesions, such as Mongolian spots, are sometimes seen in patients with mucolipidosis [180].
16.13.3 Pathogenesis
There are multiple subtypes of mucolipidoses, in which distinct genes are affected. Mucolipidosis type I is caused by deficiency in lysosomal hydrolase neuraminidase (NEU1) [152, 181, 182]. Mucolipidoses types II and III are caused by loss-of-function mutations in GNPTAB gene which encodes for the α and β subunits of the multimeric enzyme N-acetylglucosaminyl-1-phosphotransferase [183, 184]. GNPTAB enzyme catalyzes the addition of mannose-6-phosphate to lysosomal precursor proteins, and it is important for their proper trafficking from the endoplasmic reticulum to the lysosomes [185]. Interactions of mannose-6-phosphate residues on proteins with receptors on the lysosomal membrane trigger endocytosis of proteins into the lysosome. Thus, loss of GNPTAB results in defects in the trafficking of lysosomal proteins. Mutations in GNPTG gene that encodes for the γ subunit of N-acetylglucosaminyl-1-phosphotransferase are associated with mucolipidosis type III [186]. Mucolipidosis type IV is caused by mutations in TRPML1 (transient receptor potential mucolipin 1, also known as mucolipin 1 (MCOLN1) ) [187–189]. TRPML1 is a lysosomal calcium efflux channel that can affect autophagy, and its activity is regulated by the mammalian target of rapamycin (mTOR), which is a nutrient-sensitive protein kinase that negatively regulates autophagy [190].
16.14 Amyloidosis
16.14.1 Clinical Features
Localized cutaneous amyloidosis typically begins in adolescence with most pediatric cases categorized as macular, lichen, or biphasic with features of both macular and lichenoid lesions [191]. Disease usually affects individuals of Asian, Middle Eastern, and Latin American ancestry. Reported systemic associations include atopy, progressive systemic sclerosis, primary biliary cirrhosis, systemic lupus erythematosus, pachyonychia, and multiple endocrine neoplasia type 2.
Lichen amyloidosis is characterized by the coalescence of 1–10 mm skin-colored to gray or yellow, brown papules on the extensor aspects of the extremities. Some children with localized cutaneous amyloid deposits may have multiple endocrine neoplasia (MEN) type 2 syndrome, including medullary thyroid carcinoma. Lichen amyloidosis is common in these cases [199–201]. Macular amyloidosis presents as brown to gray hyperpigmented, lichenified macules with a rippled surface [191] (Fig. 16.29). Pruritus is frequently the presenting symptom in both variants. Lesions typically are persistent and progress over time. Continued trauma or rubbing may exacerbate the condition and should be discouraged.
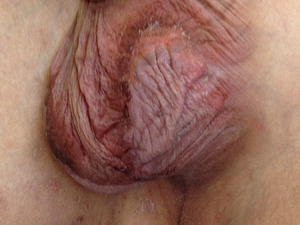
Fig. 16.29
Cutaneous macular amyloidosis presents as lichenified brown macules with subtle rippling of the skin on the scrotum of a boy (photo courtesy of Minh Hoang, MD, Ho Chi Minh City, Vietnam)
16.14.2 Histology
Cutaneous amyloidosis has several manifestations in children. Macular amyloidosis appears as small, amorphous eosinophilic deposits in the papillary dermis, often with overlying epidermal hyperpigmentation. Scattered melanophages may be seen in the papillary dermis, and a mild lymphocytic infiltrate is present in some cases [192]. This type of amyloidosis can be seen as part of X-linked pigmentary dermatosis or familial cutaneous amyloidosis [193, 194]. Some published series describe patients with this syndrome and lichen amyloidosis. The histologic findings seen in macular amyloidosis has also been described in one form of the pachyonychia congenita syndrome [195].
Lichen amyloidosis has a similar histologic appearance to macular amyloidosis, but the epidermis demonstrates lichen simplex chronicus-like changes (Fig. 16.30). Hyperkeratosis sits atop an acanthotic epidermis with hypergranulosis and elongation of the rete ridges. Eosinophilic deposits are present within the elongated papillary dermal tips (Fig. 16.31). Rare dying keratinocytes may be seen. Melanophages are present in the papillary dermis along with a scant lymphocytic infiltrate. A familial lichen amyloidosis syndrome is also described [196, 197].
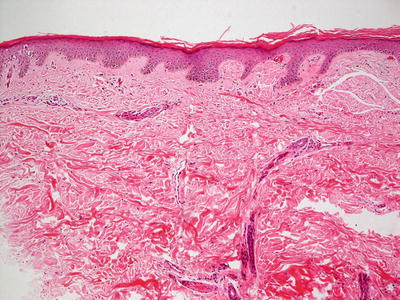
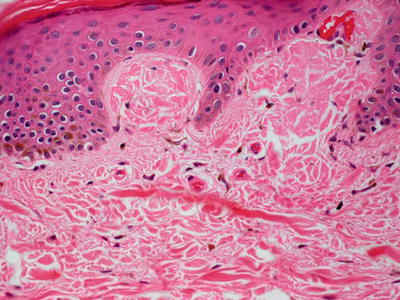
Fig. 16.30
The stratum corneum demonstrates compact orthokeratotic hyperkeratosis and the epidermis is mildly acanthotic in lichen amyloidosis
Fig. 16.31
Papillary dermal tips are expanded and filled with eosinophilic bodies (degenerating keratinocytes) in lichen amyloidosis
Regardless of the epidermal manifestations, the dermal deposits are accentuated with crystal violet stains and polarize with Congo red staining. Anti-keratin antibodies also strongly label these eosinophilic deposits. Of note, patients with these forms of amyloidosis do not have systemic gammopathies.
Nodular amyloidosis is rare in children as it is associated with plasma cell dyscrasias, which are not encountered in children with any significant frequency [198]. Nodular amyloidosis shows large acellular sheets of eosinophilic material in aggregates within the dermis (Fig. 16.32).
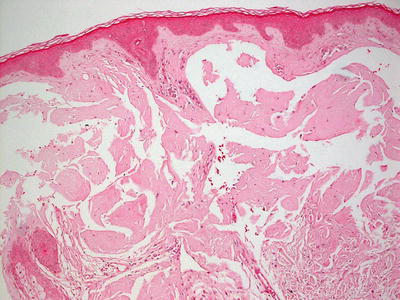
Fig. 16.32
Nodular amyloidosis demonstrates large acellular sheets of eosinophilic material in aggregates within the dermis. A characteristic “cracking” pattern is seen within the amyloid deposition
The differential diagnosis of amyloidosis includes juvenile colloid milium. The dermal material in juvenile colloid milium is believed to derive from degenerating keratinocytes [202]. It is possible that this entity is identical in its pathogenesis to lichen and macular amyloidosis, in which the amorphous eosinophilic amyloid-like material in the dermis comprises keratin breakdown products [192, 203, 204]. Lichen simplex chronicus shows identical epidermal changes to those seen in lichen amyloidosis. Close examination of the papillary dermis is essential in order to distinguish lichen simplex chronicus from lichen amyloidosis .
16.14.3 Pathogenesis
Amyloidosis is a disease of protein misfolding. It is caused by aggregation of insoluble deposits of misfolded proteins in β-pleated sheets, and the deposition of these abnormal fibrils lead to toxic effects and progressive organ dysfunction [205, 206]. Amyloid protein deposits are made up of abnormally folded serum amyloid, glycosaminoglycans and fibrillar proteins, and when bound to Congo red dye, give rise to the characteristic red-green birefringence when viewed by polarized light under the microscope [207].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


