Chapter 16 Deposition disorders
1. How is “deposition disorder” defined?
Deposition disorders comprise a diverse group of conditions or diseases in which there is accumulation, deposition, or production of substances in the skin. Typically, these substances are products of abnormal metabolism or degenerative phenomena occurring locally or systemically. The major cutaneous deposits may be subdivided into the hyalinoses, mucinoses, and mineral salts.
Touart DM, Sau P: Cutaneous deposition diseases. Part I, J Am Acad Dermatol 39:149–171, 1998.
2. What is amyloid?
Amyloid is a protein with distinct tinctorial and ultrastructural properties found as extracellular deposits. It is composed of a nonfibrillary protein known as the amyloid P component and a fibrillary component that is derived from various sources. The amyloid fibril has an antiparallel, β-pleated sheet configuration. Ultrastructurally, amyloid is composed of rigid, nonbranching fibrils measuring 6 to 10 nm in diameter.
3. How is amyloid identified?
With light microscopy, amyloid appears as amorphous, hyaline-like, eosinophilic deposits. Amyloid demonstrates green birefringence with the alkaline Congo red stain, reddish metachromasia with crystal violet, and yellow-green fluorescence with thioflavin-T stain (Fig. 16-1). These stains are not absolutely specific for amyloid, as false-positive results may occur with the other hyaline-like deposition disorders.
4. Name the various types of amyloidosis.
Amyloidosis may be classified according to clinical presentation and type of amyloid fibril protein deposition (Table 16-1). The amyloid in the macular and lichenoid variants is derived from degenerated tonofilaments of keratinocytes. Nodular amyloidosis is formed from light-chain–derived AL protein produced locally by plasma cells. It cannot be distinguished from primary systemic amyloidosis, and therefore, systemic disease should be excluded in all patients with nodular amyloidosis. There are also rare forms of hereditary systemic amyloidoses that have less frequent skin manifestations.
5. What are the cutaneous manifestations of primary or myeloma-associated systemic amyloidosis? How often do they occur?
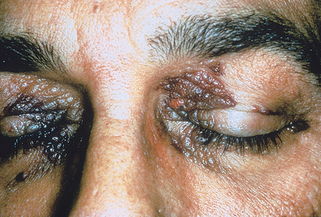
Cutaneous lesions are seen in about 30% of cases of primary or myeloma-related systemic amyloidosis. The most common skin lesions are petechiae or ecchymoses due to amyloid deposition within blood vessel walls with subsequent fragility and dermal hemorrhage. These are often seen at sites predisposed to trauma, such as the hands or intertriginous areas. Pinching the skin gives characteristic purpuric lesions known as “pinch purpura.” Purpura around the eyes may occur spontaneously but is also seen following proctoscopy or vomiting (“postproctoscopic purpura”) (Fig. 16-2). Waxy papules, nodules, or plaques may be present. Less common manifestations include sclerodermoid plaques, bullae, alopecia, and nail dystrophy.
6. Name the other organ systems that may be involved in primary or myeloma-associated amyloidosis.
Mucous membrane involvement with macroglossia occurs in 20% of cases. Hepatomegaly is found in about 50% of cases. Cardiac involvement may manifest as a restrictive cardiomyopathy or constrictive pericarditis. Peripheral nerve involvement results in paresthesias, peripheral neuropathy, and median nerve entrapment (carpal tunnel syndrome). Proteinuria is found in 80% to 90% of patients at some time during their course. Renal failure usually develops late in the disease course but may be a cause of death.
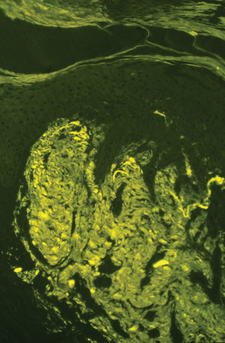
Figure 16-1. Lichen amyloidosis. Thioflavin-T demonstrates strong staining of fluorescent amyloid in the papillary dermis.
(Courtesy of James E. Fitzpatrick, MD.)
Table 16-1. Classification of Amyloidosis
| CLINICAL DISORDER | AMYLOID PROTEIN PRECURSOR | AMYLOID PROTEIN |
|---|---|---|
| Primary systemic amyloidosis | Immunoglobulin light chain | AL |
| Myeloma-associated amyloidosis | Immunoglobulin light chain | AL |
| Secondary systemic amyloidosis | Serum amyloid A lipoprotein | AA |
| Primary localized cutaneous amyloidosis | ||
| Macular amyloidosis | Keratinocyte tonofilaments | − |
| Lichen amyloidosis | Keratinocyte tonofilaments | − |
| Nodular amyloidosis | Immunoglobulin light chain (produced locally by plasma cells) | AL |
7. Compare lichen amyloidosis and macular amyloidosis.
Lichen amyloidosis is the most common form of localized cutaneous amyloidosis. Lesions are pruritic, flesh-colored to brown papules, often with overlying scale (Fig. 16-3A). Papules may coalesce into verrucous plaques. The shins are the most common site of involvement. In macular amyloidosis, pruritic macular hyperpigmentation occurs most commonly in the interscapular area. The chest or extremities are less commonly involved. The lesions have a characteristic reticulate or rippled appearance. Both of these variants of primary localized cutaneous amyloidosis occur more frequently in patients from the Middle East, Asia, and Central and South America. The etiology of both lichen and macular amyloidosis is unclear but thought to be related to chronic scratching or frictional exposure. An autosomal dominant family history may be found in up to 10% of patients with lichen amyloidosis. Lichen amyloidosis is occasionally associated with multiple endocrine neoplasia type 2A.










