Autoimmune disorders
Autoimmune lymphoproliferative syndrome (ALPS)
Immune dysregulation, polyendocrinopathy, X-linked syndrome (IPEX)
Hematologic malignancy
Acute myeloid leukemia
Acute lymphoblastic leukemia
Chronic lymphocytic leukemia
Chronic myeloid leukemia
Hodgkin’s lymphoma
Multiple myeloma
Non-Hodgkin’s lymphoma
Bone marrow failure
Aplastic anemia
Diamond-Blackfan syndrome
Fanconi anemia
Dyskeratosis congenita/Hoyeraal-Hreidarsson syndrome
Myelodysplastic syndrome
Shwachman-Diamond syndrome
Immunodeficiency
Ataxia-Telangiectasia
Chediak-Higashi syndrome
Chronic granulomatous disease
Complete interferon-γ receptor 1 deficiency
DiGeorge syndrome
DOCK8 combined immunodeficiency
Familial hemophagocytic lymphohistiocytosis
GATA2 deficiency
Griscelli syndrome
Hyper-IgM syndrome
Kostmann syndrome
Leukocyte adhesion deficiency
Severe combined immunodeficiency
Wiscott-Aldrich syndrome
X-linked proliferation syndrome
Metabolic disorders
Fucosidosis
Gaucher disease
Mucopolysaccharidoses
Osteopetrosis
Other disorders
Congenital erythropoetic porphyria (Günther disease)
Essential thrombocytopenia
Histiocytoses
Idiopathic hypereosinophilic syndrome
Myelofibrosis
Polycythemia vera
Paroxysmal nocturnal hemoglobinuria
Sickle cell disease
Thalassemia
Waldenstrom macroglobulinemia
The first step in allogeneic HSCT is the identification of a suitable stem cell donor. The donor is selected based on the similarity of his/her histocompatibility antigen (HLA) profile to that of the recipient. There are three major Class I HLA antigens, HLA-A, −B, and –C, and three major Class II HLA antigens: HLA-DP, −DQ, and –DR. Because the alleles in each HLA class tend to be inherited together, there is approximately a 25 % chance that a sibling donor will be a 6/6 HLA-identical match. If no related donor is available, then a bone marrow registry will be utilized to search for an unrelated donor. International registries now contain over 9 million potential donors and identify an unrelated donor for approximately 50 % of patients [1]. The risk of GVHD is directly proportional to the degree of mismatch in major HLA alleles between donor and recipient. In addition, mismatch of minor HLA antigens is more likely to occur in the setting of unrelated donor HSCT, also contributing to the development of GVHD. Once a suitable donor is identified, the stem cells are collected from the donor’s bone marrow, or colony-stimulating factor (CSF) is administered to the donor in order to mobilize stem cells from the marrow prior to pheresis from the peripheral blood. Umbilical cord blood is a third source for stem cell transplantation but currently accounts for a small percentage of transplants performed worldwide. The source of the stem cell graft is an important factor in the development of GVHD as peripheral blood grafts may be associated with a higher risk of GVHD than bone marrow-derived grafts [2]. After harvesting, donor stem cells are selected by physical and immunologic sorting methods. Specific T-cell depletion of the graft may be utilized in order to decrease the risk of GVHD prior to transfusion.
Pre-treatment of the recipient’s marrow before transplantation is necessary in order to allow engraftment of the donor stem cells. Traditional myeloablative regimens utilize a combination of total-body irradiation and chemotherapeutic agents such as cyclophosphamide to permit engraftment. These regimens create an immunosuppressed state, preventing the host from rejecting the foreign stem cells. Myeloablative preparative regimens may also reduce tumor burden through a direct effect on the cancer; however, they are associated with a high rate of toxicity [3]. Over the last several years, reduced-intensity (nonmyeloablative) preparative regimens have resulted in less acute toxicity and have expanded the use of allogeneic transplantation to higher risk groups including older patients or those with significant organ dysfunction. Reduced-intensity regimens rely primarily on the transplanted graft for anti-cancer activity rather than the direct cytotoxicity of the preparative regimen [1].
Autologous stem cell transplantation, which utilizes the patient’s own stem cells following ablation of the hematopoietic system, is an important treatment for certain malignancies such as non-Hodgkin’s lymphoma and multiple myeloma. More than 30,000 autologous procedures are performed worldwide each year. Although the risk of GVHD and overall non-relapse mortality are greatly reduced following autologous transplantation when compared with allogeneic transplantation, autologous procedures are associated with an increased rate of malignancy relapse [4].
Acute Versus Chronic GVHD
Traditionally, the onset of GVHD symptoms before or after the 100 day mark following transplantation has been used to designate acute versus chronic GVHD, respectively. However, this temporal distinction is somewhat arbitrary, as patients may manifest classic signs of acute GVHD after day 100, and chronic manifestations may occur before 100 days post-transplantation. Whereas acute cutaneous GVHD typically presents as an exanthematous skin eruption with gastrointestinal and hepatic involvement, chronic cutaneous GVHD is remarkable for its protean skin presentation and is associated with variable but potentially widespread organ dysfunction and immunodsyregulation. Changes in transplant protocols have also impacted the onset of acute and chronic symptoms. Nonmyeloablative conditioning regimens may delay the onset of manifestations of acute GVHD until after 100 days following transplantation [5]. Similarly, the use of donor lymphocyte infusions (DLI), wherein additional stem cells are administered weeks or months following transplantation to augment the graft-versus-tumor response, may induce symptoms of acute GVHD after the 100 day period [18].
Clinical Manifestations of GVHD
Acute GVHD
Acute GVHD is a potentially life-threatening complication of allogeneic transplantation. The risk of developing acute GVHD depends on a number of factors, including HLA-compatibility, the age and sex of the donor and recipient, the GVHD prophylaxis regimen used, and the T cell composition of the graft. Without prophylactic immunosuppression, acute GVHD will develop in most allogeneic HSCT recipients. Therefore a calcineurin inhibitor (cyclosporine or tacrolimus) or other immunosuppressant regimen is commonly used in the first several weeks to months following transplantation during which time the risk of acute GVHD is greatest.
Despite prophylactic immunosuppressive therapy, nearly 30 % of HLA-identical related transplant procedures result in significant acute GVHD [6]. This risk is significantly higher in HLA-matched unrelated and mismatched transplants. Theskin is often the earliest clinical sign of acute GVHD. Long-term survival from acute GVHD is directly related to the severity of skin, liver, and gut involvement. The 1994 Consensus Conference grading for acute GVHD is demonstrated in Table 38.2 [7].
Table 38.2
Staging and grading of acute GVHD
Stage | Skin | Liver | Gut |
|---|---|---|---|
1 | Rash <25 % BSA | Bilirubin 2 mg/dL to <3 mg/dL | Diarrhea 500–1000 mL/day or persistent nausea |
2 | Rash 25–50 % BSA | Bilirubin 3–6 mg/dL | Diarrhea 1000–1500 mL/day |
3 | Rash > 50 % BSA | Bilirubin 6–15 mg/dL | Diarrhea > 1500 mL/day |
4 | Erythroderma w/bullae formation | Bilirubin >15 mg/dL | Severe abdominal pain with or without ileus |
Grade | |||
I | Stage 1-2 | None | None |
II | Stage 3 or | Stage 1 or | Stage 1 |
III | Stege 2–3 or | Stage 2-4 | |
IV | Stage 4 or | Stage 4 |
Acute GVHD primarily involves the skin, liver, and gastrointestinal tract, although other organ systems may be affected less frequently. Skin involvement most often occurs within 2–4 weeks after transplantation. Cutaneous involvement may range in severity from an asymptomatic maculopapular erythematous eruption to widespread necrolysis, but most commonly presents with an exanthem-like eruption that preferentially involves the head, ears, palms, and soles (Fig. 38.1). In early GVHD, there may be involvement of the hair follicles creating a folliculocentric-appearance [8]. When severe, diffuse erythroderma or bullae with epidermal necrolysis may occur (Fig. 38.2).
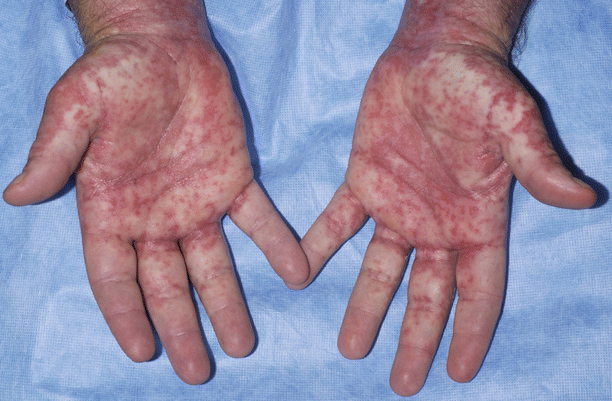
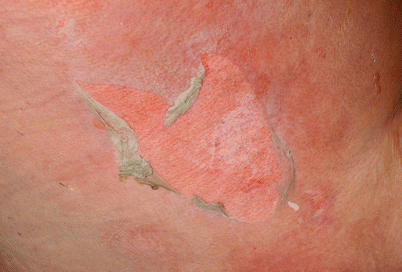
Fig. 38.1
Acute GVHD of the palms
Fig. 38.2
Acute GVHD with necrolysis
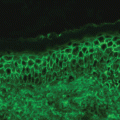
Histologically, acute GVHD is characterized by widespread keratinocyte necrosis with a dermal lymphocytic infiltrate and basal cell hydropic degeneration (Fig. 38.3). Histologic changes mimicking GVHD may be found following high-dose chemotherapy or radiation therapy, in the setting of drug hypersensitivity, and with the eruption of lymphocyte recovery; therefore clinicopathologic correlation is often helpful [9]. In cases in which the clinical and histologic diagnosis of acute skin GVHD is non-diagnostic, the presence of hyperbilirubinemia or symptoms of nausea, vomiting, diarrhea, or abdominal pain are important indicators of hepatic and gastrointestinal involvement. Even in the setting of equivocal cutaneous and histological findings, the mortality associated with severe acute GVHD necessitates a low threshold for initiating empiric corticosteroid therapy.
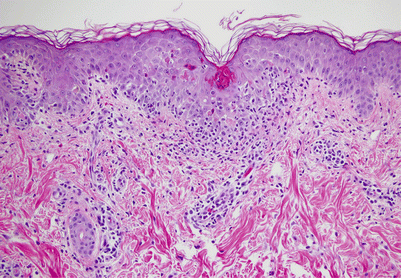
Fig. 38.3
Scattered necrotic epidermal keratinocytes with vacuolization of the basal cell layer and lymphocytic infiltration in the papillary dermis in a patient with acute GVHD (Hematoxylin and Eosin, 20×)
Chronic GVHD
Chronic GVHD occurs in 30–80 % of allogeneic HSCT recipients and is the leading cause of non-relapse mortality in survivors more than 2 years after transplantation [10]. Skin involvement may progress directly from acute disease, following a period of disease quiescence, or de novo without a history of previous acute involvement. The greatest predictor of chronic GVHD is a history of prior acute GVHD [11]. Other risk factors include older patient age, female donor for male recipient, mismatched or unrelated donor, peripheral blood graft, T-cell replete graft, and use of donor lymphocyte infusions. A flare of chronic cutaneous GVHD may be triggered by a number of factors, most commonly tapering of immunosuppression, but may also occur following the development of a drug eruption or sunburn, or in the setting of a cutaneous or systemic infection.
One of the greatest hurdles to improving chronic GVHD management stems from the clinical and immunological complexity of the disorder. In an effort to facilitate clinical research in the field of chronic GVHD, the National Institutes of Health Chronic GVHD Consensus Project published a series of articles providing a standardized approach for diagnosis and staging [12], histopathology [13], disease biomarkers [14], response criteria [15], supportive care [16], and clinical trial design [17]. Traditionally, chronic cutaneous GVHD has been described as either “lichenoid” or “sclerodermoid” involvement. However, these terms do not accurately portray the variability in the currently recognized cutaneous manifestations of chronic GVHD [18]. The Consensus Project diagnosis and staging guidelines provide a classification system of the clinical manifestations of cutaneous GVHD (Table 38.3). Poikiloderma, lichen-planus-like lesions, and sclerotic skin changes including fasciitis are considered diagnostic features of chronic cutaneous GVHD when they occur in the setting of allogeneic HSCT. Other cutaneous features, such as ichthyosis, dyspigmentation, and alopecia are also well-recognized manifestations but are not considered diagnostic of skin involvement [12].
Table 38.3
Clinical manifestations of chronic GVHD
Dermatologic and mucosal features | |
Skin | Alopecia |
Angiomatous papules | |
Bullae | |
Erythema | |
Hypo- or hyperpigmentation | |
Ichthyosis-like | |
Keratosis-pilaris-like | |
Lichen-planus-likea | |
Lichen-sclerosus-likea | |
Maculopapular | |
Morphea-likea | |
Poikilodermaa | |
Scleroderma-likea | |
Sweat impairment | |
Ulceration | |
Nails | Brittleness |
Longitudinal ridging or splitting | |
Onycholysis | |
Pterygium unguis | |
Subcutaneous tissue | Fasciitisa |
Panniculitis | |
Oral mucosa | Erythema |
Gingivitis | |
Hyperkeratotic plaquesa | |
Lichen-planus-likea | |
Mucocele | |
Mucosal atrophy | |
Mucositis | |
Pseudomembrane | |
Restriction of oral opening from sclerosisa | |
Ulcer | |
Xerostomia | |
Genital mucosa | Lichen planus-likea |
Vulvar erosions/fissures | |
Vaginal scarring/stenosisa | |
Other organ system involvement in chronic GVHD | |
Cardiovascular | Pericardial effusion |
Ophthalmologic | Cicatricial conjunctivitis |
Sicca symptoms | |
Confluent punctuate keratopathy | |
Photophobia | |
Blepharitis | |
Gastrointestinal | Esophageal web |
Esophageal stricture/stenosis | |
Hematopoeitic | Thrombocytopenia |
Eosinophilia | |
Lymphopenia | |
Hypo- or hypergammaglobulinemia | |
Autoantibodies | |
Hepatic | Elevated total bilirubin |
Elevated alkaline phosphatase | |
Elevated transaminases | |
Musculoskeletal | Myositis or polymyositis |
Edema | |
Myalgia | |
Arthralgia, arthritis | |
Neurologic | Peripheral neuropathy |
Myasthenia gravis | |
Pulmonary | Bronchiolitis obliterans ± organizing pneumonia |
Pleural effusion | |
Given the variety of epidermal changes associated with GVHD, the term “lichenoid” is preferred as a histologic descriptor of GVHD rather than as a clinical disease classification. Discrete lichen-planus-like violaceous papules are relatively uncommon in chronic GVHD, but may be seen most commonly on the palms and soles (Fig. 38.4). In the past, the term “lichenoid” has been used most commonly to describe cutaneous GVHD which manifests as poorly defined interconnecting erythematous papules and plaques with overlying scale. This eruption may localize to sites of previous UV exposure, such as the posterior and lateral neck, and may spare UV-protected areas such as the buttocks. Underlying sclerotic changes resembling lichen sclerosus or morphea may also be present. Resolution of this manifestation of chronic GVHD often results in a distinct reticulate pattern of hyperpigmentation, reflecting the pigment incontinence induced by the epidermal-dermal interface reaction.
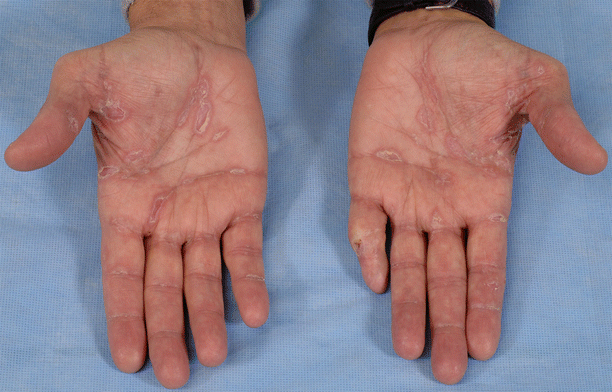
Fig. 38.4
Chronic GVHD; scaling violaceous papules and plaques on the hands
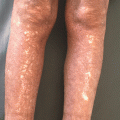
Sclerotic changes associated with chronic GVHD may develop at any layer of the dermal and subcutaneous tissue. Chosidow et al. [19] estimated the incidence of sclerodermatous GVHD to be 3.6 % based on a review of 196 HSCT patients. However, the true frequency of sclerotic changes associated with GVHD may be higher if all forms of sclerosis are included. In a large NIH cohort of 206 patients enriched for severe and refractory chronic GVHD, sclerotic changes were detected in 109 (53 %) [20]. Sclerosis may be present on multiple levels on a single patient and the changes may or may not occur in the presence of overlying epidermal involvement. The most superficial level of sclerotic changes resembles lichen sclerosus and consists of atrophic gray patches with epidermal atrophy, often distributed symmetrically on the upper back. Morphea-like GVHD mimics morphea with patchy areas of prominent dermal sclerosis, often with overlying pigmentation, that results in a decreased ability to pinch the skin. Morphea-like GVHD often occurs preferentially at sites of skin friction or pressure such as the waistband area (Fig. 38.5) [21]. Scleroderma-like GVHD represents full-thickness sclerosis with the complete inability to pinch skin and with a hidebound appearance. Involvement over joints may significantly limit range of motion. In contrast to scleroderma, scleroderma-like GVHD does not begin with symmetric distal hand involvement and proceed proximally and Raynaud’s phenomenon is uncommon [18]. Chronic sclerosis may be complicated by bullae formation as well as spontaneous erosions and ulcerations (Fig. 38.6). Benign angiomatous papules and nodules may develop in patients with chronic disease (Fig. 38.7) [22].
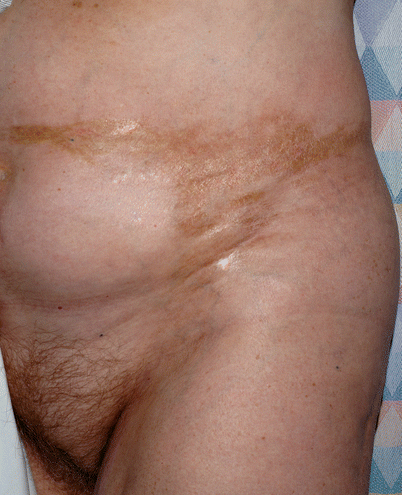
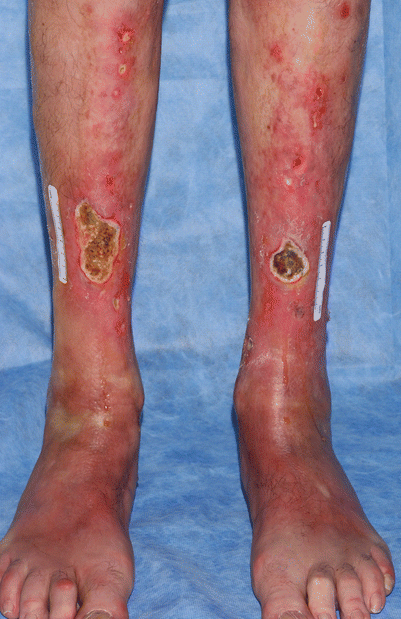
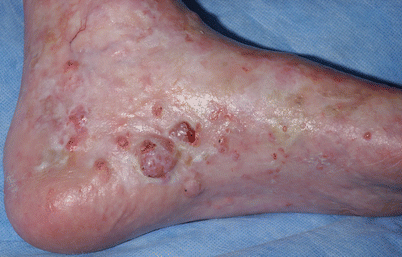
Fig. 38.5
Tan sclerotic plaques with koebnerization at site of waistband in patient morpheaform chronic GVHD
Fig. 38.6
Hidebound sclerosis with skin ulceration in a patient with scleroderma-like chronic GVHD
Fig. 38.7
Numerous angiomatous papules and nodules in a patient with chronic sclerotic GVHD of the legs
The histologic of “lichenoid” chronic GVHD resembles that of lichen planus with a bandlike lymphoplasmocytic infiltrate. Sclerotic GVHD resembles scleroderma with homogenization of collagen and loss of adnexal structures. The level and degree of fibrosis observed histologically reflects the clinical type of sclerosis. In patients with primary rippling to the skin resembling eosinophilic fasciitis, sclerosis and inflammation will be seen primarily at the interface between the reticular dermis and the subcutaneous fat and the deeper fascia.
Chronic GVHD-Related Fasciitis
GVHD-related panniculitis and fasciitis represent sclerosis of the deep subcutaneous tissue and fascia [23]. Although GVHD-related fasciitis is thought to be an uncommon presentation, it may present in an insidious manner with resultant marked functional limitations. Patients with GVHD-related fasciitis often manifest overlapping features of both panniculitis and fasciitis and the histologic diagnosis may depend on the depth and extent of the tissue biopsy. Patients may complain of muscle pain or weakness [24], or may demonstrate limited range of motion at affected joints at the time of presentation. GVHD-associated fasciitis shares many similarities with eosinophilic fasciitis, an uncommon disorder of unknown etiology first described by Shulman in 1974 [25]. GVHD-associated fasciitis presents with prominent induration and rippling of tissue, visible grooves demarcating fascial bundles or underlying superficial veins, and decreased range of motion (Fig. 38.8). The first indications of subcutaneous involvement may be edema of the affected limb. Magnetic resonance imaging may facilitate the diagnosis of subcutaneous involvement [26, 27].
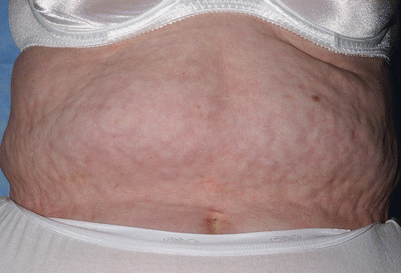
Fig. 38.8
Subcutaneous sclerosis from chronic GVHD; there is prominent rippling and nodularity of the subcutaneous tissue appreciable by deep palpation. The overlying skin is normal in texture and color
Genital GVHD
Assessment of genital involvement is important in the management of GVHD patients. Genital involvement is most commonly associated with sclerotic cutaneous disease, but may occur with other forms of cutaneous involvement, or in the absence of other cutaneous involvement. Genital tract involvement may be present in as many as 49 % of female patients two years post-transplantation and may seriously impact the quality of life of affected individuals [28]. Manifestations include burning and irritation, discharge, erosions and fissures, or vaginal stricture (Fig. 38.9). In a recent review of 155 male allogeneic transplant recipients 1 year or more after transplant, 21 (13 %) manifested inflammatory lesions, most frequently balanoposthitis (12 patients), lichen-sclerosus-like lesions (6), and phimosis (5) [29].
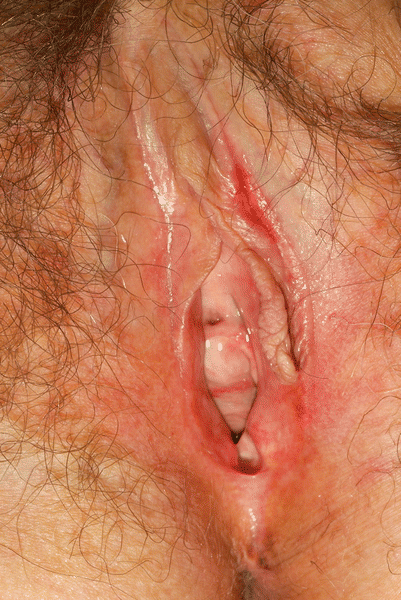
Fig. 38.9
Chronic vulvar GVHD; resorption of labia minora; pallor and sclerosis of vulvar vestibule; fissuring of the interlabial sulcus
Oral GVHD
The second most common organ system involved with chronic GVHD after the skin is the mouth. Chronic oral GVHD may affect the oral mucosa and salivary glands. Mucocele formation is very common but usually asymptomatic. Lichen-planus-like oral involvement manifests as erythematous hyperkeratotic plaques and erosions. Sclerosis of the skin surrounding the mouth or the frenulum can cause difficulty opening the mouth or protruding the tongue, respectively. Persistent erosions and fissures cause burning pain, particularly upon contact with acidic foods. Salivary gland involvement from chronic GVHD results in decreased saliva production and sicca symptioms. Loss of taste is also commonly reported by patients [30].
Immunology of GVHD
Acute GVHD
Acute GVHD is a reaction of immunocompetent donor cells against the cells and organs of the host. Billingham [31] described three features necessary for the development of GVHD: (1) the transplanted graft must be immunologically competent; (2) the recipient must not be capable of rejecting the graft; (3) the recipient must express antigens that are recognized as foreign by the graft. Grafted cells recognize the host as foreign through differences between the donor and host in major and minor HLA expression.
Pro-inflammatory Environment
Ferrara and Reddy [32] proposed a three-step model for the immunopathophysiology of acute GVHD. The first phase occurs prior to transplantation of the graft during which time chemoradiotherapy, the underlying disease state, and other factors activate host antigen presenting cells (APCs). Total body irradiation in particular plays an important role in priming the immune response by inducing epithelial cell damage in the gastrointestinal tract, which leads to host secretion of inflammatory cytokines (TNF-α and IL-1) and exposure to microbial products such as lipopolysaccharide. In fact, total body irradiation may contribute to chronic GVHD as well, particularly sclerotic skin disease, as pre-transplant conditioning was recently found to a be a risk factor for development of sclerotic chronic GVHD [20].
In the second phase of acute GVHD, host APCs expressing MHC class I and II molecules are recognized as foreign by donor T-cells. In murine studies, host dendritic cells are sufficient to activate donor T cells [33]. Differences in minor histocompatibility antigens (mHags), such as those encoded on the Y chromosome, may also play an important role in propagating acute GVHD, particularly in the setting of HLA-identical transplantation. Activation of natural killer (NK) cells, which eliminate APCs, abrogates the development of GVHD, suggesting a crucial role for host APCs in the propagation of acute GVHD [34]. The three major organ systems involved in acute GVHD – the skin, liver, and gut – contain large numbers of APC, which may in part be responsible for the localization of tissue damage to these organ systems [35].
In the final effector phase, inflammatory mediators and cell-mediated killing work together to induce the clinical effects typical of acute GVHD. CD8+ cytotoxic T cells (CTLs) utilize perforin/granzyme mediated cytolysis, whereas CD4+ T cells utilize Fas/FasL signaling, which may be particularly important for inducing hepatic damage [32]. TNF-α and IL-1 signaling play a prominent role in cellular damage in acute gastrointestinal GVHD. Cytokine gene polymorphisms may influence the expression of GVHD as a variance in the TNF-α gene has been associated with an increased risk of severe acute GHVD [36], whereas polymorphisms in IL-10, a potent suppressor of TNF-α, IL-1, and other inflammatory cytokines, has been associated with a decreased incidence of acute GVHD [37].
Regulatory T-Cells
Acute GVHD is mediated by donor T-cells that expand following transplantation in response to the recipient environment. Regulatory T-cells (Treg) constitute a subset of the T-cell population which exert control over the allogeneic T-cell response against the host. Tregs express FOXP3, a key transcription factor for Treg function, as well as CD25, the IL-2 receptor α chain that is also expressed by activated T-cells. Donor grafts with a low percentage of CD4 + Foxp3+ Tregs are associated with an increased risk of acute GVHD. In addition, the ratio of CD4 + FOXP3+ cells to CD4 + FOXP3-CD25+ in patients after transplant is significantly reduced in patients with acute GVHD, suggesting an important role for Tregs in control of effector function [38]. Manipulation of specific T-cell subsets in donor grafts may allow for modulation of the GVHD and graft-versus-leukemia response.
Chronic GVHD
In contrast to acute disease, our understanding of the pathogenesis of chronic GVHD is somewhat incomplete. Chronic GVHD demonstrates a complex interplay of immunologic processes with features of alloimmunity, autoimmunity, and immunodeficiency. The heterogeneity of clinical manifestations and disease course in chronic GVHD makes identification of a murine model challenging. In addition, correlations between murine and human chronic GVHD are difficult because marked differences in immune reactions are observed by differences in the intensity of the conditioning regimen, disparity between strains, donor graft composition, and endogenous microbes of the animals [39]. The best characterized murine model of GVHD is the parent-into-F1 mouse. In this model, parental lymphocytes are injected into the F1 recipient offspring. Because the parental lymphocytes are genetically related to the recipient, they are not recognized as foreign. However, the donor lymphocytes recognize the F1 mouse as foreign, inducing a GVHD reaction.
In chronic GVHD, autoreactive T-cells are thought to arise from impairment of negative thymic selection due to thymic damage incurred from chemotherapy, acute GVHD, or age-related atrophy [40]. The clinical similarity of chronic GVHD to autoimmune diseases such as Sjogren’s syndrome and scleroderma and the reported benefit of treatment of chronic GVHD with anti-CD20 monoclonal antibody [41] also suggest a role for humoral immunity in chronic GVHD. Circulating autoantibodies were described in one of the first series describing the clinical features of chronic GVHD in 1980 [42]; however, the relevance of antibody formation to disease activity remains unclear. In a prospective study, the cumulative incidence of antinuclear antibodies (ANA) in patients with extensive chronic GVHD was 94 % after a median follow-up time of 26 months. The presence of nucleolar pattern ANA and other antibodies in association with ANA was associated with an increased risk of extensive chronic GVHD [43]. However, in this study, the presence and titer of specific autoantibodies did not predict the type of organ involvement, a finding in accord with a recent study comparing 106 patients with sclerotic GVHD to patients with GVHD without skin sclerosis which failed to find an association with ANA or other scleroderma-related antibodies [20, 43]. Nevertheless, antibodies targeting minor-HLA antibodies on the Y chromosome in male recipients of stem cell grafts from female donors correlate with the presence of chronic GVHD, suggesting a potentially important interplay between T and B cells in the pathogenesis of chronic GVHD [44].
Cytokine Dysregulation
Chronic GVHD is associated with elevated levels of IL-1 (β), IL-6, transforming growth-factor-β, TNF-α, and IFN-γ as well as decreased levels of IL-10 [45, 46]. TGF-β is the cytokine that has been most strongly implicated in GVHD-related fibrosis. Tissue fibrosis is a common manifestation in several organ systems involved with chronic GVHD, including the liver, pulmonary system (bronchiolitis obliterans), and skin. TGF-β is a pleiotropic cytokine that in the acute post-transplant period regulates donor engraftment and graft-versus-leukemia effect [47]. In the chronic period, TGF-β appears to the major driving force for collagen synthesis and the development of fibrosis. In the murine sclerodermatous GVH model, treatment with anti-TGF-β antibody prevented lung and skin fibrosis [48].
Platelet Derived Growth Factor
In scleroderma, platelet-derived growth factor (PDGF) appears to play a key role in the increased proliferative capacity of fibroblasts, an effect which is enhanced by the presence of transforming growth factor-β [49]. Increased gene expression of PGDF has been also detected in the skin in the murine sclerodermatous GVHD model [50]. Baroni et al. [51] reported stimulatory autoantibodies to the platelet-derived growth factor receptor (PDGFR) in a group of 46 patients with systemic sclerosis. Ten additional patients with scleroderma-like GVHD were also reported to have agonistic antibodies (this group was not further described in the paper). In this study, production of reactive oxygen species (ROS) and tyrosine phosphorylation was reversed with the use of PDGFR tyrosine kinase inhibitors, suggesting that agents such as imatinib mesylate with activity against the PDGFR may have potential utility for targeting this signaling pathway in the setting of scleroderma and sclerotic GVHD. However, to date the identification of stimulatory PDGFR antibodies in systemic sclerosis or GVHD has not subsequently been confirmed by other groups.
Donor Lymphocyte Infusion and Graft-Versus-Tumor Effect
An important barrier to the effective control of acute and chronic GVHD is the risk of inhibiting the activity of the stem cell graft against the donor’s primary malignancy (graft vs. leukemia effect). Numerous studies have demonstrated that the risk of tumor relapse is lower in patients with GVHD than in those that do not develop GVHD [52]. Similarly, T-cell depletion of the donor graft and aggressive multi-agent GVHD prophylaxis reduces the risk of developing GVHD, but does so at the expense of an antileukemic effect, resulting in an increased relapse rate [53]. Donor lymphocyte infusions (DLI) have been utilized to augment the anti-tumor response of the graft but are also associated with an increased rate of GVHD [54]. Ideally, our understanding of chronic GVHD will progress to the point where separation of the graft vs. host and graft vs. leukemia (tumor) effect would be possible. In reality, it is a constant struggle to balance these competing forces in the management of these patients.
Treatment of Cutaneous GVHD
Acute GVHD
Acute GVHD is treated with systemic steroids, resulting in a 40–50 % response rate. There is no consensus as to the appropriate second-line agent in those patients who do not respond adequately to corticosteroid therapy. A variety of salvage therapies have been utilized in patients with steroid refractory GVHD, but no single agent has proven to be a superior option (Table 38.4) [110]. The major limitation of most acute GVHD therapies arises from the use of systemic immunosuppression and attendant risk of infection. Newer biological agents, such as those targeting tumor necrosis factor-α, have shown some benefit in acute GVHD but have been linked to invasive fungal infections [111].
Table 38.4
Treatment for acute and chronic mucocutaneous GVHD
Treatment | Type of GVHD | |
|---|---|---|
Acute | Chronic (L/Sc/Oral) | |
Antithymocyte globulin | Remberger et al. [55] | – |
Azathioprine | – | Penas et al. [56] (Sc) |
Epstein et al. [57] (Oral) | ||
Basiliximab | Funke et al. [58] | – |
Clofazimine: | – | Lee et al. [59] (L/Sc) |
Corticosteroids (systemic) | Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |