(1)
Department of Dermatology, University of Pennsylvania, Penn Presbyterian Medical Center Medical Arts Building, Philadelphia, PA, USA
18.2 Basement Membrane
18.3 Cornified Envelope
18.5.2 Important Oncogenes
18.5.3 Genes in Melanoma
18.5.8 Defects in Nuclear Envelope
18.5.9 Defects in DNA Repair Genes
18.6 Embryology
18.7 Melanocyte Biology
18.8 Nail Biology
18.9 Sebum
18.10 Hair
18.11 Itch Review
18.12 Immunology Review
18.12.1 Innate Immune Response
18.12.2 Adaptive Immune Response
Abstract
This section reviews high yield facts behind the most important areas of basic science relevant to dermatology.
Keywords
Basic scienceGenes18.1 Keratinocyte Adhesion
Occluding:
Tight junctions (zonula occludens)
Transmembrane proteins: claudins and occludins (cytoskeletal component: actin)
Intracellular proteins: e.g. ZO-1, ZO-2, ZO-3 (zonula occludins)
Anchoring:
Desmosomes (macula adherens): “slow, strong”
Desmosomal plaque (Fig. 18.1) = Intermediate filament → desmoplakin → plakoglobin → desmosomal cadherins (transmembrane proteins) = desmocollin, desmoglein (desmosomes connected by homo- and hetero-dimers of cadherins)
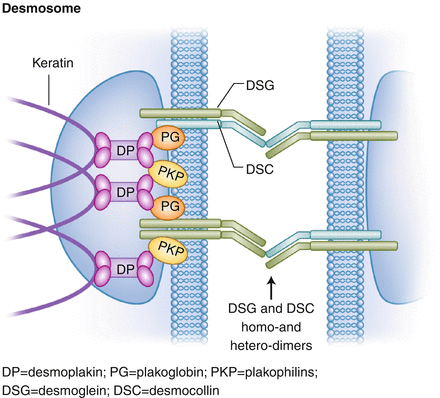
Figure 18.1
Components and structure of the desmosome. DP desmoplakin, PG plakoglobin, PKP plakophilins, DSG desmoglein, DSC desmocollin
Intermediate filaments = keratin in epithelia/skin (desmin in muscle, vimentin in fibroblasts and endothelial cells)
Cadherins = calcium-dependent adhesion transmembrane proteins, includes both desmocollin and desmoglein
Dsg1 = more in upper layers of epidermis, less in mucosae; Ab causes pemphigus foliaceus, also Staph exfoliative toxin A (Staph group 2, phage 71) against Dsg1 causes SSSS and bullous impetigo. Genetic defect in striate PPK. Homozygous mutant may cause severe dermatitis, allergies (to food), and metabolic wasting (SAM syndrome)
Dsg2 = mostly in heart and simple epithelia (e.g. gut), small amount in skin and increased expression in SCC
Dsg3 = more in lower layers of epidermis, but all throughout mucous membranes (Ab causes pemphigus vulgaris)
Dsg4 = mostly in hair follicles, mutated in AR monilethrix
Desmocollin-1 = Ab causes IgA pemphigus (subcorneal pustular dermatosis)
Desmocollin-2 = mutation can cause R-sided cardiomyopathy along with mild PPK and woolly hair
Desmocollin-3 = mutation associated with hypotrichosis and vesicles
Desmoplakin = defect causes Carvajal syndrome (Woolly hair, L- sided cardiomyopathy, epidermolytic PPK) also linked to striate PPK and lethal acantholytic epidermolysis bullosa
Plakoglobin = defect causes Naxos syndrome (Woolly hair, R- sided cardiomyopathy, PPK); can also cause lethal congenital EB, but only a couple case reports
Plakophilin-1 = defect causes ectodermal dysplasia-skin fragility (McGrath) syndrome
Adherens junctions (zonula adherens): “quick, weak”
E-cadherin attached to actin via α- and β-catenin
E-cadherin = defect in gastric cancer, E-cadherin also expressed by Langerhans cells to allow homing to epidermis
β-catenin = mutated in pilomatricomas; inactivation of APC in Gardner’s causes β-catenin accumulation and pilomatricoma-like changes in associated EICs
Communicating:
Gap junctions (nexus)
Six connexins form connexon
Two connexons (two hemichannels) form gap junction
Connexins preferentially expressed in inner ear/cornea, epidermis and follicular unit (affects hearing, hair/skin)
Connexin 26 = defect causes Vohwinkel’s syndrome (with deafness), KID (keratitis, ichthyosis, deafness) syndrome, PPK with deafness, Bart-Pumphrey syndrome
Connexin 30 = defect causes hidrotic ectodermal dysplasia (Clouston’s syndrome)
Connexin 30.3, 31 = defect causes erythrokeratodermia variabilis
18.2 Basement Membrane
The structure of the basement membrane zone (BMZ) contains many components (Fig. 18.2).
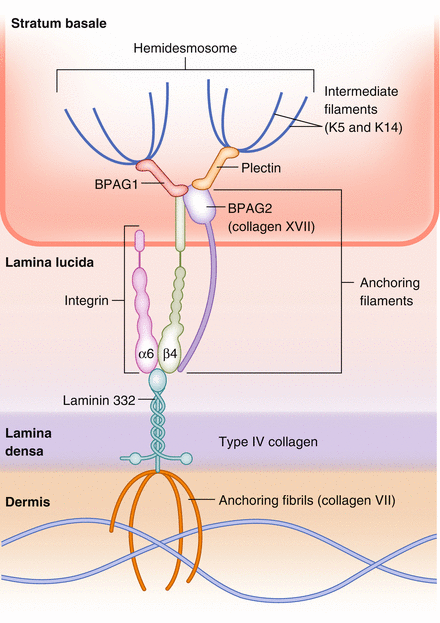
Figure 18.2
Components and structure of the basement membrane zone (dermal-epidermal junction)
Note: the hemidesmosome (at the BMZ) and the desmosome (keratinocyte to keratinocyte adhesion) share similarities, but are different. The BMZ can be divided into 4 zones:
Zone 1 – plasma membrane and hemidesmosome (keratin 5 and 14) = basal keratinocyte, plectin/BPAG1
Zone 2 – lamina lucida (cleavage plane of salt-split skin) = BPAG2, α6β4 integrin (anchoring filaments) [interface between lamina lucida and densa = laminin-5/332/epiligrin = binds anchoring filaments (BPAG2 and α6β4 integrin) to anchoring fibrils (NC1 domain of type VII collagen)]
Zone 3 – lamina densa (basement membrane proper) = type IV collagen, heparan sulfate, nidogen (BMZ)
Zone 4 – sublamina densa (papillary dermis) = anchoring fibrils, type VII collagen
Internal plaque (inside basal keratinocyte) = plectin, BPAG1 (230 kD) (plakin family); plectin (also a plakin) primarily binds β4 of α6β4, BPAG1 binds BPAG2 and β4; both BPAG1 and plectin bind to keratins as well to link the cytoskeleton to the hemidesmosome
External plaque = BPAG2 (180 kD), integrins (α6β4) = anchoring filaments
Internal plaque (plectin, BPAG1) connects with external plaque at α6β4 integrin connecting to BPAG2 at NC16A region to α6.
Laminin-332 (aka laminin-5 or epiligrin) connects between external plaque anchoring filaments (α6β4) and lamina densa (type IV collagen)
Keratins (intermediate filaments), K5 and K14 in basal cells
Type VII collagen comprises the anchoring fibrils
Defect in keratin 5 and 14 = EB simplex
Defect in keratin 5 = EB simplex with mottled pigmentation, Dowling-Degos, Galli-Galli
Defect in keratin 14 = dermatopathia pigmentosa reticularis (DPR) and Naegeli- Franceschetti-Jadassohn syndrome (NFJS)
Defect in laminin-5 = junctional EB
Defect in plectin = EB with muscular dystrophy
Defect in α6β4-integrin = JEB with pyloric atresia
Defect in type VII collagen (anchoring fibrils) = dystrophic EB
Ab to BPAG1, 2 = bullous pemphigoid
Ab to laminin-5 (“anti-epiligrin”) = cicatricial pemphigoid (Brunsting-Perry [limited to head/neck] and mucosal membrane type); can be associated with underlying malignancy
Ab to BPAG2 = bullous pemphigoid, herpes gestationis, cicatricial pemphigoid (C-terminal portion), linear IgA bullous dermatosis (97 kD segment)
Ab to α6β4-integrin (specifically β4) = ocular-specific cicatricial pemphigoid
Ab to BPAG2 (97 kD segment), NC16A of BPAG2 = linear IgA
Ab to type VII collagen = EB acquisita, bullous lupus
Note: the “plakin family” includes BPAG1 (BP230), plectin, desmoplakin, envoplakin, periplakin (may see Ab against any or all in paraneoplastic pemphigus)
Plakoglobin (a catenin) = may be part of both desmosome and adherens junctions
18.3 Cornified Envelope
Keratinocytes mature up the epidermis and ultimately form the stratum corneum/cornified envelope (Fig. 18.3).
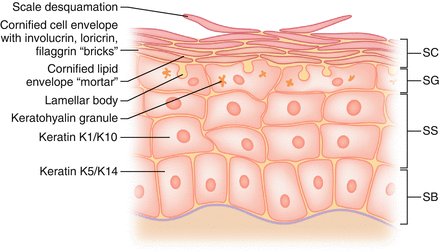
Figure 18.3
Keratinocyte maturation and development of the cornified envelope
Principal mechanical barrier of the skin = stratum corneum
Important in regulation of water release (TEWL = trans-epidermal water loss)
Bricks and mortar model = bricks (corneocytes or protein envelope), mortar (lipid envelope = extracellular lipid matrix from lamellar bodies)
Cornified envelope consists of protein envelope and lipid envelope
Granular layer contains keratohyalin granules = involucrin, loricrin (70–85% of corneum mass), profilaggrin (proteins), which become the main components of the cornified envelope
Profilaggrin → filaggrin, proteins cross-linked by transglutaminases
Corneocytes formed from keratinocytes losing their lipids (that become the lipid envelope lamellar bodies), leaving the proteins (filaggrin, loricrin) behind
Odland bodies (lamellar bodies, keratinosomes) made in spinous layer, make ceramides, contain glycolipids; these become the lipid components of the envelope
In final stage of keratinocyte differentiation (from basal layer maturing up), keratins are aligned into arrays by filaggrin (FILament AGGRegating), and cross-linked by transglutaminases
As filaggrin matures up stratum corneum, dehydration triggers filaggrin degradation by cathepsins
Defect in cathepsin-C = Papillon-Lefevre and Haim-Munk syndromes
Defect in filaggrin = ichthyosis vulgaris
Defect in loricrin = Vohwinkel syndrome with ichthyosis (no deafness), NBCIE
Defect in transglutaminase-1 = lamellar ichthyosis, congenital ichthyosiform erythroderma (spectrum of disease includes both)
Defect in steroid sulfatase = X-linked ichthyosis
Defect in ABCA12 = Harlequin fetus (can also be seen in lamellar ichthyosis)
Defect in phytanoyl-CoA hydoxylase (PEX7) = Refsum disease
Defect in fatty aldehyde dehydrogenase = Sjögren-Larsson
Defect in LEKTI (encoded by SPINK-5 (Serine Protease INhibitor)) = Netherton’s
Defect in corneodesmosin (component of desmosomes in the stratum corneum) = hypotrichosis simplex
Defect in Odland bodies (lamellar bodies) = in Flegel’s disease (hyperkeratosis lenticularis perstans)
Ab to transglutaminase-3 = dermatitis herpetiformis
Keratins:
Acidic keratins (type I) (9–20), basic keratins (type II) (1–8)
Keratin 1 and 10 = suprabasal, Keratin 5 and 14 = basal layer,
Keratin 9 (acidic) = only expressed in suprabasal palmar/plantar (partners with K1, basic)
Defect in keratin 1 and 10 = epidermolytic ichthyos is (aka bullous congenital ichthyosiform erythroderma (BCIE) or epidermolytic hyperkeratosis (EHK))
Defect in keratin 1 and 9 = Vorner (epidermolytic PPK) → in palmoplantar skin, 1 and 9 pair (K10 not expressed in palmoplantar)
Defect in keratin 1 = Unna-Thost (nonepidermolytic); Mnemonic: Unna = Uno = 1
Defect in keratin 2e = ichthyosis bullosa of Siemens
Defect in keratin 3 and 12 = Meesmann corneal dystrophy
Defect in keratin 4 and 13 = white sponge nevus (buccal mucosa)
Defect in keratin 5 and 14 = EB simplex
Defect in keratin 5 alone = Dowling-Degos, Galli-Galli
Defect in K6a and 16 = pachyonychia congenita type 1
Defect in K6b and 17 = pachyonychia congenita type 2 (steatocystomas)
Defect in K14 (alone) = Naegeli-Franceschetti-Jadassohn syndrome (NFJS), dermatopathia pigmentosa reticularis (DPR)
18.4 Extracellular Matrix (ECM)
Composed of network of collagens, elastin, glycoproteins, and proteoglycans
Collagen types
Collagen I = dermis in skin (most of ECM) and bone
Collagen II = cartilage
Collagen III = dermis in skin (especially fetal skin), vasculature, wound healing (primary component of granulation tissue)
Collagen IV = basement membrane
Collagen V = defect in classic Ehlers-Danlos syndrome
Collagen VII = anchoring fibrils
Collagen XVII = BPAG2 transmembrane protein/anchoring filaments
Collagen biosynthesis
Collagen molecules synthesized in endoplasmic reticulum (ER); then they are assembled into triple helices composed of three polypeptide chains (α-chains) via hydroxylation (by vitamin C dependent prolyl and lysyl hydroxylases)
Procollagen triple helix is secreted from ER across Golgi into extracellular space; it is then cleaved and assembled/cross-linked into various fibrils/superstructures (using enzymes including copper-dependent lysyl oxidase)
Extracellular steps include cleavage and assembly into superstructures
Enzymes involved in biosynthesis: prolyl hydroxylases and lysyl hydroxylase; these require Fe and ascorbic acid as co-factors
Cross-linking between collagen molecules is catalyzed by lysyl oxidase, which requires copper as a co-factor
Elastin
Elastic fibers composed of the protein elastin surrounded by mesh of microfibrils; number one component = fibrillin, which may guide elastogenesis and add structural support. Fibulin is another microfibril- associated component.
Elastic fibers cross-linked by desmosine and isodesmosine via lysyl oxidase (the same copper-dependent enzyme that cross-links collagen)
Elastin gene one of the largest in the human genome
Two types of elastin fibers:
Elaunin = in reticular dermis, parallel
Oxytalan = in papillary dermis, perpendicular
Embryology
Embryonic dermis watery/cellular, ratio of collagen III: collagen I = 3:1 (opposite of adult dermis which is more collagen I). Fetal skin does not scar until this ratio changes, also related to TGF-ß isoforms
Inherited collagen/elastin disease
Defect in fibrillin-1 = Marfan syndrome
Defect in fibrillin-2 = congenital contractural arachnodactyly
Defect in fibulins 4 or 5 = cutis laxa (Note: Defect in folliculin = Birt-Hogg-Dubé syndrome)
Defect in collagen I = osteogenesis imperfecta (OI)
Defect in collagen V, Tenascin X = EDS I/II (Classic)
Defect in collagen III (COL3A1), Tenascin X = EDS III (Hypermobility)
Defect in collagen III (COL3A1) (α1-chain of type III collagen) = EDS IV (Vascular)
Defect in lysyl hydroxylase = EDS Type VI (Kyphoscoliosis)
Defect in collagen I (COL1A1,1A2) (α1 and α2 chains type I collagen) = EDS Type VII (Arthrochalasia)
Defect in ADAMTS-2 (procollagen N-proteinase) = EDS type VIIC (dermatosparaxis)
Defect in collagen type VII = dystrophic EB
Defect in ABCC6 (ATP-dependent binding cassette) = pseudoxanthoma elasticum (PXE)
Defect in ECM-1 = lipoid proteinosis
Defect in collagen IV = Alport’s syndrome
Defects in copper metabolism: ATP7A (Menkes kinky hair) and ATP7B (Wilson’s)
Autoimmune collagen disease
Ab to collagen IV = Goodpasture syndrome (glomerulonephritis, pneumonitis)
Ab to collagen II = relapsing polychondritis and MAGIC syndrome (mouth and genital ulcers with inflamed cartilage)
Ab to collagen XVII (BPAG2) and laminin-5 = bullous pemphigoid and cicatricial pemphigoid
Ab to collagen VII = EBA, bullous lupus
Ab to ECM-1 = lichen sclerosus
Penicillamine
Copper chelator
Interferes with elastin cross-linking (1. By causing low copper blood levels that cause reduced lysyl oxidase activity, 2. By directly blocking cross-linking of residues)
Used to treat Wilson’s disease
Can cause PXE, EPS, acquired cutis laxa, pemphigus foliaceus-like drug eruption
Collagen as filler
Semi-permanent → Bovine collagen (Zyderm)
Permanent fillers → Polymethylmethacrylate beads suspended in bovine collagen (Artefill/Bellafill)
18.5 Molecular Biology Review
Molecular techniques
Polymerase chain reaction (PCR) = amplifies/copies a gene or specific sequence of DNA using specific primers, DNA polymerase (e.g. Taq polymerase), and nucleotides through repeated thermal cycles
DNA sequencing = determines exact sequence of nucleotides in a gene
Western blot = detects protein with labeled antibodies
Southern blot = detects DNA with radioactive DNA probe
Northern blot = detects mRNA with radioactive DNA probe
DNA microarray = scans an entire genome for gene expression
Other terminology
Chromosome: composed of long arms (q) and short arms (p) = “petite”, connected by centromere (site of microtubule attachment = kinetochore)
Tumor suppressor gene = a gene that protects a cell from uncontrolled proliferation; loss-of-function mutation leads to cancer
Proto-oncogene = a gene that if mutated (gain-of-function mutation) can become an oncogene and promote uncontrolled proliferation, leading to cancerRelated posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



