(1)
Department of Dermatology, Cleveland Clinic Foundation, Cleveland, OH, USA
(2)
Department of Dermatology, Division of Dermatologic Surgery, Wilmot Cancer Center, University of Rochester, 400 Red Creek Drive, Suite 200, Rochester, NY 14623, USA
Introduction
Basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) are the two most common types of cutaneous neoplasms [1]. The worldwide incidence of nonmelanoma skin cancer (NMSC) has increased by 3–8% every year, and the incidence in the US may increase by 50% by the year 2030 [2–4]. In order to treat these tumors, especially in their aggressive forms not amenable to surgery, an understanding of their pathogenesis is integral. A multitude of factors interact with one another to promote tumorigenesis and tumor survival. This chapter will discuss the pathogenesis of BCC and SCC, with particular emphasis on how aberrant angiogenesis promotes tumor progression, and will elucidate nonsurgical treatment modalities, both old and new, that directly or indirectly target angiogenesis in their anti-proliferative actions.
Basal Cell Carcinoma
Pathogenesis
BCC is the most common skin cancer, comprising about 80% of NMSC [5]. BCC arises from the cells of the basal layer of the epidermis and hair follicles [6]. The pathogenesis and proliferation of this tumor is a multifactorial process that eventually leads to cellular proliferation and differentiation, tumor survival, and angiogenesis (Fig. 4.1, Table 4.1). In recent years, cancer genetics have elucidated the role epidermal growth factor receptor (EGFR) activation in the development of BCC [7]. EGFR appears to be mutated, dysregulated, or over-expressed in many cancers, including BCC [7]. As a result, mutated or over-expressed EGFR leads to impaired immune response in human skin and promotion of cellular proliferation, differentiation, and survival [8].
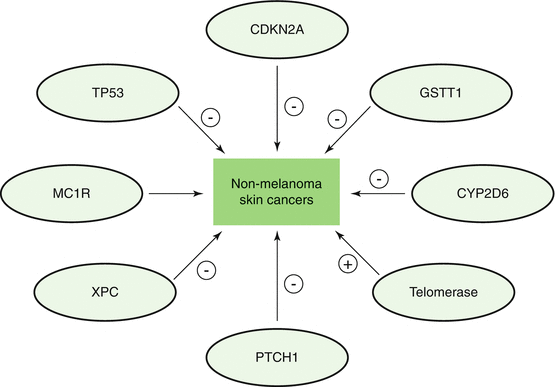
Fig. 4.1
Major pathways involved in the pathogenesis of non-melanoma skin cancers. CDKN2A cyclin-dependent kinase inhibitor 2A, GSTT1 glutathione S-transferase theta 1, CYP2D6 cytochrome P450, family 2, subfamily D, polypeptide 6, PTCH1 patched homolog 1, XPC xeroderma pigmentosum, complementation group C, MC1R melanocortin 1 receptor, TP53 tumour protein 53 (Adapted with permission from Madan et al. [47])
Table 4.1
Environmental risk factors for non-melanoma skin cancers
Type of non-melanoma skin cancer | |
|---|---|
Solar UV radiation | BCC, SCC |
Human papillomavirus | SCC, BCC |
Iatrogenic immunosuppression | SCC, BCC |
HIV/AIDS and non-Hodgkin lymphoma | BCC, SCC |
PUVA therapy | SCC, BCC |
Photosensitising drugs (e.g., fluoroquinolone antibiotics) | SCC, BCC |
UVB radiation | BCC |
Ionising radiation | BCC |
Occupational factors | BCC, SCC |
Arsenic | SCC, BCC |
Tobacco smoking | SCC |
In addition to EGFR signaling, established research has shown that the Hedgehog (Hh) signaling pathway is the predominant player in the tumorigenesis of BCC (Fig. 4.2) [7]. The Hh pathway is quiescent and nonfunctional in mature skin. It is composed of patched-1 (PTCH), a transmembrane receptor, which normally exerts an inhibitory effect on smoothened (SMO), a transmembrane protein. When PTCH is dysfunctional, it cannot apply its normal inhibitory effect on SMO. In its unregulated active form, SMO promotes cellular replication and ultimately, tumor development. The majority of BCC carry a mutation in the PTCH-1 resulting in inactive PTCH-1, while a minority carry a mutation in the SMO gene resulting in overexpression of the Hh pathway [7].
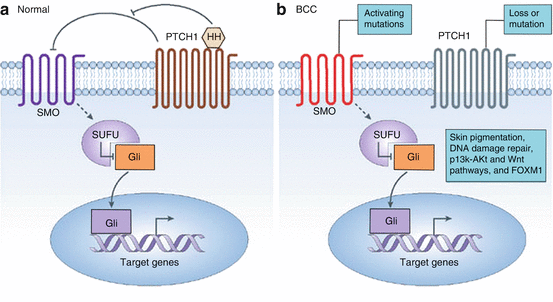
Fig. 4.2
The Hedgehog (HH) signaling pathway. (a) Normally, patched 1 (PTCH 1) inhibits smoothened (SMO). Sonic hedgehog (SHH) can bind to the PTCH 1 receptor, thereby relieving the inhibition of SMO by PTCH1; SMO then sends signals through a series of interacting proteins, including suppressor of fused (SUFU), resulting in activation of the downstream Gli family of transcription factors (GLI1, GLI2 and GLI3). (b) Sporadic BCCs routinely carry mutations in PTCH1 and TP53, and, in 10% of instances, in SMO. Other mutations implicated in BCC development include genes that regulate skin color, DNA damage repair genes, members of the phosphoinositide 3-kinase (PI3K)–Akt and the Wnt pathways and FOXM1 (Adapted with permission from: Epstein [48])
In addition to promoting the proliferation and differentiation of adult stem cells, the Hh signaling pathway has also been shown to be a key mediator of angiogenesis and likely tumor survival [9]. In early embryonic development, the Hh pathway plays a normal role in angiogenesis. It has been shown that this pathway induces the expression of vascular endothelial growth factors (VEGFs) and migration of endothelial cells [9]. The Hh pathway also leads to a signaling cascade that ultimately aids in coronary and pulmonary angiogenesis [10, 11]. In post-natal life, reports have shown that in ischemic or injured tissue, Hh expression is increased, playing a role in regeneration, repair, and survival [12]. Therefore in an aberrant, over-expressed state such as in BCC, Hh signaling may lead to unregulated angiogenesis and tumor survival [13].
Treatment
Although surgical excision remains the gold standard for the treatment of BCC, patients who are poor surgical candidates or with metastatic BCC may not be amenable to surgical intervention. Therefore, research has brought to the forefront several alternative modalities in the treatment of BCC that among many other mechanisms, retard tumor growth, oftentimes acting on its angiogenic properties. The most exciting development in the treatment of BCC has been the development of oral Hh inhibitors. The first of these to come to market, vismodegib (Erivedge, Genentech Roche) was recently approved for the treatment of locally advanced or metastatic disease. As previously mentioned, when PTCH-1 is dysfunctional, it fails to exert its normal inhibitory effect on SMO, leading to overexpression of SMO and cellular proliferation (Fig. 4.2). Vismoedgib specifically acts as a SMO inhibitor, thus acting at a downstream point in the Hh pathway and having efficacy whether tumors harbor mutations in PTCH-1 or SMO. In a Phase I trial of 33 patients with refractory advanced or metastasized BCC, there was an overall 58% response, either partial or complete, and the duration of response was 12.8 months and ongoing; several reports have shown similar results [14]. Other well-established drugs have been experimentally employed in the treatment of BCC, such as the EGFR-inhibitor cetuximab. In one case report, an 87-year-old man with giant, nodular mid-facial BCC showed minimal progression over 4 months with treatment with cetuximab [15]. In another case of two palliative patients, tumor stability was noted with cetuximab, and one patient developed liver metastases following cessation of treatment [16]. Finally, a case series reported that when four patients with BCC were treated with cetuximab, two had complete remission and two had partial remission; three patients relapsed after treatment cessation [17].
In addition to systemic therapy, several topical modalities have been trialed in the treatment of BCC. 5-fluorouracil (5-FU) 5% cream is approved by the US Food and Drug Administration (FDA) clearance rate in lesions after twice a day application for 8–12 weeks [18]. In addition to 5-FU, imiquimod, a topical immunomodulator, has been successfully used in the treatment of BCC. Imiquimod 5.0% cream is US FDA approved to treat nonfacial superficial BCC [19]. Once daily application, 5-days per week for 6–12 weeks leads to the highest clearance rate of lesions (up to 80%) [18]. This topical drug activates toll-like receptor 7 (TLR 7) [19]. TLR 7 is subsequently responsible for the upregulation of many different cytokines, including interferon-α (IFN-α), tumor necrosis factor-α (TNF-α) and interleukin-12 (IL-12) (Fig. 4.3) [20]. The role of Type 1 interferons (IFNs) in the apoptosis and anti-angiogenesis of BCC cannot be understated.
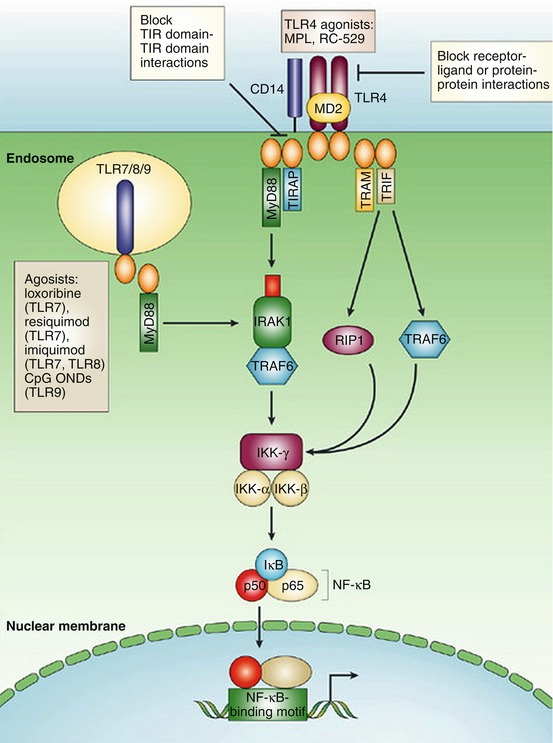
Fig. 4.3
Imiquimod signaling pathway, as part of a complex interation of toll-like receptors (TLRs) and the immune system. Imiquimod acts upon TLR7 within the cellular endosome, which in turn interacts with a cytoplasmic adaptor receptor, MyD88. MyD88 mediates the association with the serine-threonine kinase, IRAK. This in turn leads to the ultimate activation of nuclear factor-κΒ (NF-κΒ) and synthesis of numerous Type I interferons (Adapted with permission from Ulevitch [49])
IFNs are naturally occurring glycoproteins that are secreted by cells in response to biologic stimuli, including viral infection [21]. Since their discovery, research has elucidated their anti-viral, anti-proliferative, and immunomodulatory properties [22]. Type I IFNs (IFN-α/β) have been shown to have an anti-proliferative, prodifferentiation effect on normal keratinocytes. Keratinocytes supplemented with Type 1 IFNs exhibited an average of 70% growth inhibition, as well as terminal differentiation. Several reports have demonstrated the effect of Type 1 IFNs on antiangiogenesis. Mouse IFNs α/β have been shown to impair wound healing in mice by inhibiting the endothelial and epidermal cell proliferation [23]. Another study showed that mice implanted with IFN α/β and proangiogenic factors such as VEGF demonstrated a significantly less number of blood vessels than the mice without IFN α/β [24]. Moreover, the immunomodulatory effects of Type 1 IFNs have been explained by experiments that have demonstrated the induction of class I major histocompatibility (MCH) antigen expression in keratinocytes treated with Type 1 IFNs [25]. Through these innovations, the mechanism by which imiquimod may play a role in the destruction of BCC has been elucidated.
In addition, IFN-α has been explored as a direct treatment for BCC. One group demonstrated that IFN-α demonstrated growth inhibitory effects on primary BCC cell lines in vitro [26]. In another study of 15 patients with BCC, treatment with IFN-α led to the expression of CD95 and CD95L, which ultimately led to cell death by suicide and fratricide [27]. In one report, four patients with nodular BCC were treated with intralesional IFN-α and demonstrated resolution of the BCC on histopathologic examination [28]. A similar study demonstrated that in six patients with nodular and superficial BCC treated with intralesional IFN-α, two BCCs were cured and four showed clinical and histologic signs of improvement [29]. Given its anti-proliferative, anti-angiogenic, and immunomodulatory properties, IFN-α therapy may be a plausible option in the treatment of patients who are poor surgical candidates, or in order to shrink tumor size prior to surgery.
Squamous Cell Carcinoma
Pathogenesis
SCC is the second most common cutaneous neoplasm, after BCC. As in the development of BCC, the pathogenesis of SCC is a multifactorial and multistage process that occurs gradually over time in response to chronic ultra-violet (UV) radiation exposure (Fig. 4.1 , Table 4.1). In 1928, Findlay was the first to describe that chronic UV radiation could induce SCC in mice [30]. During the initial phases of UV-induced damage, keratinocytes undergo point mutations. Nearly 60% of SCC demonstrates mutations in the p53 tumor suppressor gene [31]. UV-induced mutations in p53 impair its normal ability to activate DNA repair enzymes, arrest the cell cycle, and induce cellular apoptosis, leading to aberrant cellular proliferation and tumor development [32]. Once tumorigenesis has initiated, a multitude of factors interplay to support tumor growth and survival. One such key factor in the development of SCC, and to a lesser extent BCC, is the increased expression of the enzyme cyclooxygenase-2 (COX-2) in the epidermis. COX-2 is integral in the formation of prostaglandins, potent mediators of inflammation, angiogenesis, and immunosuppression (Figs. 4.4 and 4.5) [33]. In preclinical studies, mice that were deficient in COX-2 exhibited significantly less numbers of SCC than wild-type mice, and in a clinical trial by Elmets and colleagues, patients treated with celecoxib, a COX-2 inhibitor, exhibited significantly fewer NMSC than patients treated with placebo [34, 35]. Furthermore, the formation of reactive oxygen intermediates promotes DNA damage and tumor progression [32]. Finally, as in BCC, SCC, especially of the head and neck, demonstrate overexpression of EGFR, leading to signal transduction that induces carcinogenesis [36].
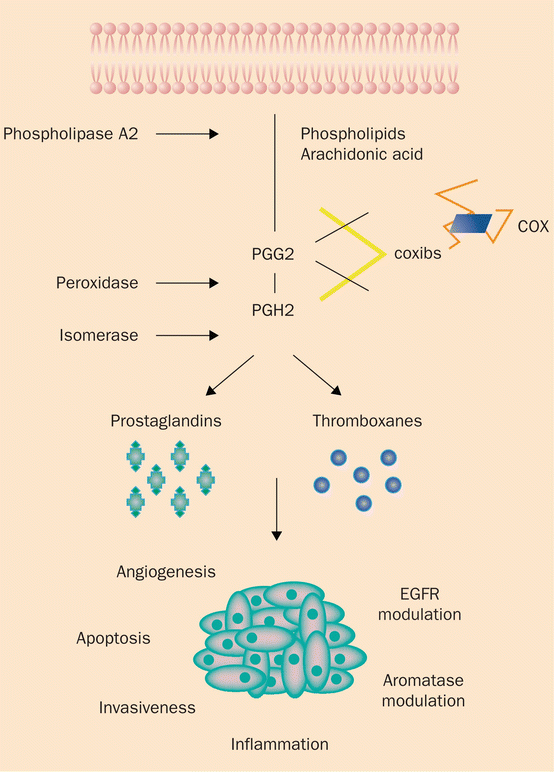
Fig. 4.4
 Infantile Hemangioma: New Insights on Pathogenesis and Beta Blockers Mechanisms of Action
Infantile Hemangioma: New Insights on Pathogenesis and Beta Blockers Mechanisms of Action
 Angiogenesis: General Concepts
Angiogenesis: General Concepts
 Chemoprevention and Angiogenesis
Chemoprevention and Angiogenesis
 The Role of Angiogenesis in the Development of Psoriasis
The Role of Angiogenesis in the Development of Psoriasis
 Angiogenesis and Pathogenesis of Port Wine Stain and Infantile Hemangiomas
Angiogenesis and Pathogenesis of Port Wine Stain and Infantile Hemangiomas
 Potential Role of Angiogenesis and Lymphangiogenesis in Atopic Dermatitis: Evidence from Human Studies and Lessons from an Animal Model of Human Disease
Potential Role of Angiogenesis and Lymphangiogenesis in Atopic Dermatitis: Evidence from Human Studies and Lessons from an Animal Model of Human Disease




COX catalyzes the conversion of arachidonic acid to prostaglandins, and ultimately angiogenesis and tumor progression. COX cyclooxygenase, PGG2 prostaglandin G2, PGH2 prostaglandin H2, EGFR epidermal growth factor receptor. (Adapted with permission from Gasparini et al. [50])
Related posts:
Infantile Hemangioma: New Insights on Pathogenesis and Beta Blockers Mechanisms of Action
Angiogenesis: General Concepts
Chemoprevention and Angiogenesis
The Role of Angiogenesis in the Development of Psoriasis
Angiogenesis and Pathogenesis of Port Wine Stain and Infantile Hemangiomas
Potential Role of Angiogenesis and Lymphangiogenesis in Atopic Dermatitis: Evidence from Human Studies and Lessons from an Animal Model of Human Disease
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
