Fig. 9.1
Cancer incidence in the small intestine, colon and rectum increases with age. (a) Intestinal cancer is the second biggest cancer threat for human beings after lung cancer. (b) The colon and rectum are much more prone to cancer development, whereas the small intestine seems to be more protected (The data is retrieved from the Center for Disease Control WONDER database (http://wonder.cdc.gov) and covers the United States cancer statistics from 1999 until 2004)
The examples above indicate that a young, healthy organism is able to respond to a variety of external stresses with different magnitudes. The young are able to balance dietary starvation, ischemia/reperfusion damage or irradiation within limits and bring about equilibrium, whereas the old encounter complications much earlier. All processes mentioned are influenced to various degrees by the intestinal epithelium and its renewal capacity suggesting that the dynamic range of appropriate stress responses becomes narrower upon old age. The reasons for these age-associated changes can be speculated upon and may partially be explained by stem cell dysfunction due to damage accumulation.
9.3 Intestinal Stem Cells
Epithelial stem cells in the small intestine reside at the bottom of narrow invaginations coined as the crypts of Lieberkühn. Dividing stem cells give rise to highly proliferative progenitor cells, so-called transit amplifying cells, which finally start to differentiate, producing absorptive and secretory cells (van der Flier and Clevers 2009). Differentiated cells are mainly located in epithelial extensions called villi where absorptive enterocytes support further breakdown of the food with subsequent nutrient uptake. Secretory goblet cells produce mucous, Paneth cells are involved in host defence, and enteroendocrine cells secrete hormones regulating digestion, blood flow and intestinal motility. Proliferation at the crypt base and apoptosis at the villus tip in combination with a cellular upward flow create a conveyor belt along the crypt-villus axis, which allows epithelial renewal in a matter of around 5 days (Fig. 9.2). The hierarchical organization of the colon is very similar to the small intestine with the exception that neither villi nor Paneth cells are generated.
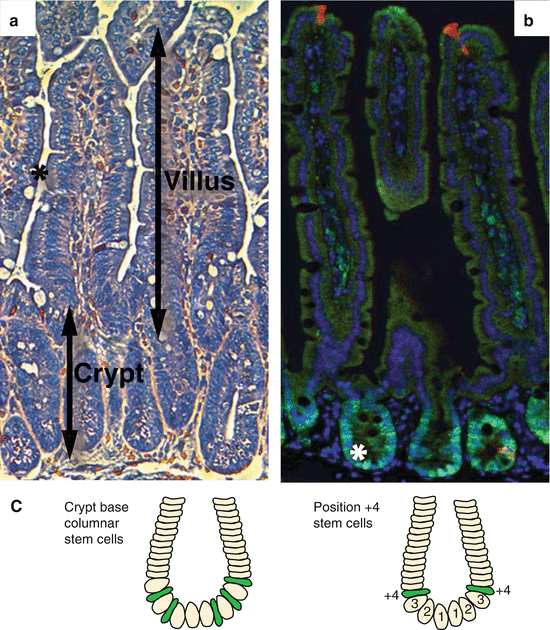
Fig. 9.2
Architecture of the small intestine. (a) Haematoxylin- and eosin-stained small intestine section. The small intestine is composed of luminal villi and basal crypts. The outer villus epithelium is composed of differentiated enterocytes, mucous secreting goblet cells (asterisk) and few enteroendocrine cells. Each villus is furthermore connected to several crypts containing proliferating cells and granular Paneth cells at the crypt bottom. (b) Small intestine section labelled against PCNA (green), cleaved caspase 3 (red) and DNA (blue). Basal crypts of Lieberkühn contain dividing stem and transit-amplifying cells and stain positive for the proliferation marker PCNA. The crypt base columnar stem cells (asterisk) are readily visible as cone-shaped cells at the base of the crypt interspersed with nondividing Paneth cells (label only blue). Shedding of exhausted cells at the tip of the villus is executed by apoptosis indicated by cleavage of caspase 3 (red). (c) Location of stem cells according to the stem cell zone (left) and the position +4 model (right)
Recent years gained further insight into stem cell biology of the gastrointestinal tract harnessing mouse models, which unambiguously allowed defining stem cells based on genetic marker expression and lineage-tracing experiments. Lineage tracing employs restricted Cre recombinase expression in the cell type in question and fate mapping of all genetically labelled cellular progeny produced by this cell: epithelial intestinal stem cells by definition are able to produce all differentiated cell types and persistently keep the label due to self-renewal divisions (Snippert and Clevers 2011). One of the markers for intestinal stem cells identified by these means is the seven-transmembrane receptor Lgr5 (leucine-rich repeat-containing G-protein-coupled receptor 5), which is specifically expressed in highly proliferative crypt base columnar (CBC) stem cells in the small intestine and the colon. Other experiments functionally demonstrated Bmi1- (Sangiorgi and Capecchi 2008), Tert- (Montgomery et al. 2011), and Hopx-expressing cells (Takeda et al. 2011) to be intestinal stem cells residing at the crypt position ‘+4’, i.e. directly above Lgr5-expressing CBC stem cells. The ‘+4’ cells are more quiescent than the proliferative CBC cells and elegant experiments demonstrated an interconversion between the cycling CBC state and the more quiescent ‘+4’ state (Takeda et al. 2011; Montgomery et al. 2011; Tian et al. 2011; Yan et al. 2012). It has to be noted though, that the quiescent cells originally proposed to exclusively occupy crypt position ‘+4’ may be more variable with regard to their location (Yan et al. 2012; Itzkovitz et al. 2012). These data in combination with the notion that Bmi1 is restricted to the small intestine prompted the search for markers of quiescent intestinal stem cells in the colon and led to the discovery of a subset of crypt cells expressing the EGF receptor inhibitor Lrig1 (leucine-rich repeats and immunoglobulin-like domains 1) (Powell et al. 2012; Wong et al. 2012). Lrig1 is expressed in the colon and small intestine and labels quiescent stem cells that can be activated to proliferate upon challenge by stress like irradiation (Powell et al. 2012). In summary, the data suggest the existence of several stem cell states, which can be assigned in order to react to changing conditions or upcoming challenges.
Within the proliferating cell fraction, intestinal stem cells are more sensitive to ionizing irradiation than the transit-amplifying cells (Barker et al. 2007; Hua et al. 2012; Potten 1977). But it is questionable whether increased damage sensitivity would really be advantageous at the tissue level since total stem cell depletion would ultimately lead to a loss of the critical cell compartment that is required for tissue repair. In fact, recent experiments revealed an exception by demonstrating a rapid loss of Lgr5-expressing crypt base columnar stem cells upon irradiation, which is followed by a brief proliferative expansion of the Bmi1 ‘+4’ cells. The activated Bmi1 population was suggested to subsequently give rise to new cycling Lgr5-positive crypt base columnar stem cells and afterwards re-enter the quiescent state (Yan et al. 2012). In line with this data was the demonstration that Dll1 (delta-like 1)-expressing secretory progenitor cells are also able to revert to cycling Lgr5-expressing stem cells after irradiation with ionizing radiation (van Es et al. 2012). These and other analyses reveal a cellular reserve that can be called upon under conditions of stress in order to preserve organ maintenance (Tian et al. 2011; Itzkovitz et al. 2012). The evolving concept encompasses irradiation-sensitive cycling intestinal stem cells and quiescent irradiation-resistant stem cells, which become temporarily activated in order to regenerate an irradiation injury. It will be interesting to determine if the stem cell composition is altered during the aging process and if such alteration may be involved in the changes of stress response evident at old age.
9.4 Stem Cell Protection and DNA Damage Accumulation
Different types of molecular damage affecting proteins, RNA, DNA, mitochondria and other structures accumulate during aging. There is evidence that DNA damage contributes to stem cell and organismal aging: (1) aging-dependent accumulation of DNA damage is detected in various tissues of mammals including the intestine (Dimri et al. 1995; Herbig et al. 2006; Hasty et al. 2003; Lombard et al. 2005; Jiang et al. 2007; Dolle et al. 2000). The age-dependent accumulation of DNA damage also affects stem and progenitor cells in the intestine (Rossi et al. 2007; Rube et al. 2011; Hewitt et al. 2012; Wang et al. 2009). (2) Enforced regeneration during chronic disease is associated with the accumulation of telomere dysfunction and DNA damage, leading to regenerative exhaustion, disease progression and organ failure, for example, in ulcerative colitis (O’Sullivan et al. 2002). DNA damage is not the primary cause of these diseases, but it accumulates as a consequence of elevated rates of cell turnover, and the accumulation of DNA damage leads to disease progression. (3) Intestinal stem cells express telomerase and suffer telomere length shortening with increasing age (Schepers et al. 2011). (4) Studies in genetically engineered mice with defective telomere maintenance and DNA repair capacity provided functional proof for the causal relation between DNA damage accrual and intestinal dysfunction, leading to accelerated aging (Rudolph et al. 1999; Murga et al. 2009; de Boer et al. 2002; Niedernhofer et al. 2006).
Given the highly proliferative renewal of the intestinal epithelium in the face of potentially noxious and pathogenic species (Garrett et al. 2010), the maintenance of functional stem cells appears to be of utmost importance to ensure functional integrity of the intestine during aging. Several mechanisms could be involved in protecting intestinal stem cells from damage accumulation: (1) intestinal stem cells reside in an anatomically protected site, the crypt, which is even further occluded by copious amounts of mucous, greatly reducing exposure to potentially toxic luminal content. Paneth cells at the crypt bottom additionally secrete bactericidal components (Bevins and Salzman 2011). (2) The ABC transporter P-glycoprotein/MDR1 is transiently upregulated in the crypt and villus during regeneration after ischemia-reperfusion damage (Omae et al. 2005) though it is not vital for basic function (Schinkel et al. 1997). The activity of a multidrug-resistant protein like MDR1 is able to generate a so-called side population in fluorescence-activated cell sorting. Putative stem cells from the small intestine and colon have been isolated as a side population, but their stem cell identity was not unambiguously proven at the time (Dekaney et al. 2005; Samuel et al. 2009). Stem cells in various organ systems and cancer stem cells have been shown to express high levels of multidrug-resistant proteins (i.e. ATP-binding cassette – ABC transporters), likely increasing the resistance of stem cells to toxic substances and preventing damage accumulation (Dean et al. 2005; Visvader and Lindeman 2008). (3) There is evidence that an asymmetric separation of damaged proteins occurs during stem cell division, keeping intestinal stem cells free of damaged proteins (Rujano et al. 2006). Such mechanism of asymmetric segregation of proteins has also been described in yeast cell division in a way that the mother will keep damaged proteins away from the daughter cell (Shcheprova et al. 2008). It is tempting to speculate that this mechanism is also active in the intestine to prevent damage accumulation in long-living stem cells. (4) Asymmetric DNA strand segregation was furthermore suggested to support genomic integrity of long-lived stem cells by maintaining an immortal, unreplicated DNA strand in stem cells to protect these against replication-induced DNA mutations (Cairns 1975; Potten et al. 1978, 2002; Quyn et al. 2010). This concept has recently been challenged by several studies arguing for a random strand separation (Escobar et al. 2011; Schepers et al. 2011; Steinhauser et al. 2012) thus leaving it open if stem cells are able to modify DNA strand separation under certain conditions. (5) In line with the latter data indicating a symmetric distribution of DNA templates are findings revealing a symmetric stem cell division in the intestinal crypt (Lopez-Garcia et al. 2010; Snippert et al. 2010). Furthermore, the crypt is populated by equipotent stem cells that neutrally compete for space at the crypt base. Cell division and cell loss support a clonal drift in each crypt eventually giving rise to a clone. Stem cell populations with reduced fitness could thus easily be replaced by more healthy stem cells supporting organ maintenance during periods of stress and aging. On the other hand, damaged cells with a growth advantage could also take over and substitute the undamaged stem cell population leading to organ dysfunction and aging or to the formation of malignancies like cancer. In this light, it is clearly necessary to prevent damage accumulation via checkpoint responses in order to preserve proper organ maintenance and function.
9.4.1 Checkpoint Responses in Intestinal Stem Cells
Studies on genetically engineered irradiated rodents have shed light on the mechanisms protecting the intestine from DNA damage accumulation and organ failure. The acute survival of mice after increasing doses of gamma irradiation is determined by the function of either the haematopoietic system or the gastrointestinal tract. Doses of around 12 Gy cause death in a matter of weeks due to failure of the haematopoietic system (Lorenz et al. 1951), whereas higher doses of around 15–18 Gy lead to death after some days due to intestinal failure (Sangiorgi and Capecchi 2008; Hua et al. 2012) suggesting some kind of protection of the intestinal stem cells up to a certain irradiation threshold.
This protection is partially mediated by a tight regulation of apoptosis and proliferation at stem- and progenitor level. On the one hand, stem and progenitor cells are protected from immediate obliteration by irradiation-induced DNA damage by expression of anti-apoptotic genes Bcl2 and Bcl-W (Bcl2l2). Mice knockout for either of the two factors suffer substantially more irradiation-induced apoptosis (Merritt et al. 1995; Pritchard et al. 2000; Pritchard et al. 1999). Interestingly, expression of Bcl2 is higher in the colon than in the small intestine, and the levels of spontaneous apoptosis rise only in the colon but not in the small intestine of Bcl2 knockout mice (Merritt et al. 1995). These findings are in line with the reduced apoptotic response in the colon compared to the small intestine (Wilson et al. 1998) indicating a higher damage tolerance or improved repair mechanisms.
On the other hand, the proliferating stem and progenitor cells display a higher sensitivity towards ionizing radiation than differentiated cells (Potten 1977; Wilson et al. 1998; Hua et al. 2012) and commence apoptosis induced by the pro-apoptotic p53 target gene PUMA (p53-upregulated modulator of apoptosis) (Qiu et al. 2008). The transcription factor p53 has previously been implicated to mediate the apoptotic response upon irradiation since p53 knockout mice were partially protected from irradiation-induced cell death (Merritt et al. 1994, 1997). This data suggests that organ maintenance is preserved by apoptotic removal of very damaged cells and demands that a surviving fraction of stem cells must be sustained as well.
Proliferation control via cell cycle arrest or senescence induction is another way of preserving organ homeostasis to allow DNA repair or permanently suppress cell division of a damaged cell. Key components of the DNA repair machinery are the protein kinases ATM and ATR, which are activated by DNA double- and single-strand breaks, respectively (Shiloh 2003; Cimprich and Cortez 2008). The protein kinases Chk1 and Chk2 are ATM/ATR targets, which slow down cell cycle progression by reducing cyclin-dependent kinase (CDK) activity mediated by activation of the transcription factor p53 and its targets, e.g. the CDK inhibitor p21 (Kastan and Bartek 2004). Checkpoint induction may increase the time available for proper DNA repair before replication or mitosis. Deletion of ATR or Chk1 leads to rapid stem cell depletion, which is probably linked to the essential role of these kinases in S phase progression of replicating cells (Cimprich 2007; Ruzankina et al. 2007; Greenow et al. 2009). However, it is currently not known whether ATR and Chk1 contribute to the regulation of stem cell self-renewal and quiescence. In case the damage cannot be repaired, chronic checkpoint signalling triggers either cell death or permanent cell cycle arrest, i.e. senescence (Campisi and d’Adda di Fagagna 2007).
Critically short telomeres tend to fuse, induce fusion-bridge-breakage cycles and cause chronic DNA damage thus inducing permanent cell cycle arrest (O’Sullivan and Karlseder 2010; Shay et al. 1991). Generally, it has been demonstrated that intestinal stem cells are relatively resistant to radiation by quickly engaging homologous recombination and non-homologous end-joining DNA repair machineries thus restoring genomic integrity faster than transit-amplifying cells (Hua et al. 2012).
There is some evidence on CDK inhibitors and components of the DNA damage response to regulate intestinal stem and progenitor cells. Deletion of the senescence inducer p21 allows more stem and progenitor cells to proliferate under steady-state conditions and improves survival after irradiation (George et al. 2009). In aging mice with dysfunctional telomeres, p21 has been shown to limit self-renewal of intestinal stem cells, and co-deletion of p21 extends organ maintenance (Choudhury et al. 2007). The CDK inhibitors p27 and p57 have also been implicated to regulate intestinal stem and progenitor cell behaviour by regulating cell cycle progression and affecting differentiation. Upregulation of both factors upon deletion of Notch receptors causes loss of proliferating cells and a dramatic increase in differentiation into goblet cells (Riccio et al. 2008). Age-related changes in CDK inhibitor expression may thus impact on organ homeostasis through control of stem cell self-renewal via proliferation and differentiation surveillance.
Studies on senescent rodents and elderly humans have revealed changes in basal and stress-induced levels of apoptosis and proliferation. Elderly humans display with increased basal levels of apoptosis and proliferation in the small intestine (Ciccocioppo et al. 2002; Corazza et al. 1998). Starvation and re-feeding of senescent rats are associated with increased proliferation and decreased apoptosis compared to young animals (Holt et al. 1988; Xiao et al. 2001). Elevated Akt signalling has been implicated to mediate this effect by inhibiting pro-apoptotic Bad and releasing anti-apoptotic Bcl-XL (Bcl2l1) in aging rat colon (Majumdar and Du 2006). Moreover, irradiation of senescent mice revealed a stronger apoptotic response and a growth delay compared to younger mice (Martin et al. 1998a, b). These data clearly demonstrate age-related changes in cellular stress response pathways and may contribute to the lowered stress response range in older individuals. The underlying mechanistic changes and the cell populations affected are far from being understood, though.
9.5 Checkpoints Lead to Depletion of Damaged Stem Cells
As previously outlined, checkpoint genes affect intestinal stem cells under conditions of stress and during normal homeostasis. Because DNA damage contributes to limitations in stem cell functionality and tissue homeostasis during aging, it is of special interest to study the influence of checkpoint genes in this context. Given the aging-associated decline of telomere length and telomere-associated DNA damage in intestinal stem cells (Schepers et al. 2011; Hewitt et al. 2012), Terc knockout mice with dysfunctional telomeres represent a valuable tool to study the role of checkpoint genes in DNA damage-driven aging (Rudolph et al. 1999). Studies in these aging telomere dysfunctional mice revealed some insight on the function of checkpoint genes influencing premature aging (Fig. 9.3). Interestingly, the reduced organ maintenance in mice with short telomeres is the result of impairments in stem cells, e.g. in the colon or small intestine (Rudolph et al. 1999).
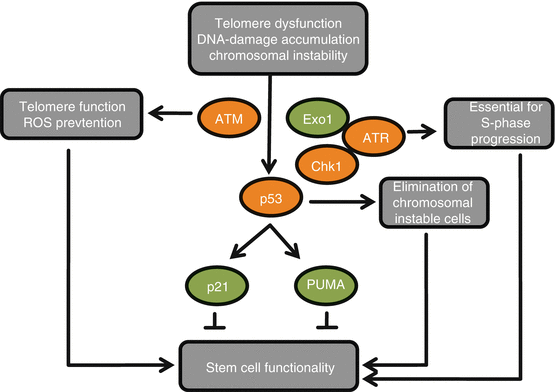
Fig. 9.3
Deletions of checkpoint genes influence DNA damage-induced impairment of somatic stem cells. Telomere dysfunction and other types of DNA damage lead to activation of the p53 pathway. In this context, deletions of checkpoint genes have positive (green) or negative (red) effects on stem cell functionality. Loss of transcriptional targets of p53 (such as p21 or PUMA (p53-upregulated modulator of apoptosis)) increases functionality and tissue maintenance. By contrast, the deletion of p53 itself leads to chromosomal instability of stem cells and accelerated tissue aging. The p53-dependent depletion of genetically instable stem cells is tissue protective. Upstream of p53 and deletion of ataxia telangiectasia mutated (ATM) accelerate stem cell dysfunction by increasing telomere dysfunction. Impairment of exonuclease 1 (Exo1)-mediated activation of ATR inhibits p53-dependent DNA damage responses and prolongs stem cell function without inducing chromosomal instability at the stem cell level. Although not directly tested in telomere-dysfunctional mice, deletion of ATR or its downstream kinase checkpoint kinase 1 (Chk1) is expected to have immediate adverse effects owing to its essential role in S phase progression
9.5.1 Effects of p53 Deletion in Telomere-Dysfunctional Mice
p53 deletion in telomere-dysfunctional intestinal epithelium results in improved maintenance but chromosomal instability of intestinal stem cells, leading to defects in stem cell differentiation and accelerated atrophy of the intestinal epithelium (Begus-Nahrmann et al. 2009). These results indicate that p53 has a tissue-protective effect by removing chromosomally instable stem cells from aging somatic tissues. Decreased responsiveness of ATM–p53-dependent checkpoints occurs in aging mouse tissues (Feng et al. 2007). It is possible that this age-dependent loss of p53 checkpoint function contributes to the acceleration of tissue atrophy during aging.
9.5.2 Effects of Altered Expression of p53 Target Genes
Telomere dysfunction and irradiation-induced DNA damage also lead to increased expression of p53 target genes in somatic stem cells (Choudhury et al. 2007; Sperka et al. 2012a). In the context of telomere dysfunction, upregulation of p21 expression is associated with decreased self-renewal and functionality of stem cells, and deletion of p21 significantly rescues both stem cell self-renewal and their ability to maintain tissue integrity in the intestinal epithelium (Choudhury et al. 2007).
Telomere dysfunction also leads to an upregulation of PUMA (p53-upregulated modulator of apoptosis), a downstream target of p53 that is required for p53-dependent depletion of adult stem cells (Liu et al. 2010). Similarly to p53 deletion, PUMA deletion rescued the maintenance of intestinal stem cells in aging telomere-dysfunctional mice (Sperka et al. 2012a). However, in contrast to p53 deletion, PUMA deletion did not result in chromosomal instability of intestinal stem cells but prolonged the maintenance of the intestinal epithelium of telomere-dysfunctional mice (Sperka et al. 2012a). Cell culture studies on freshly isolated intestinal stem cells revealed that PUMA-dependent apoptosis and p21-dependent cell cycle arrest cooperate in limiting self-renewal of adult stem cells in response to telomere dysfunction. Deletion of either one of the checkpoint branches results in prolonged self-renewal of intestinal stem cells without inducing chromosomal instability. Simultaneous impairment of both checkpoint branches leads to a further increase in the self-renewal capacity of cultured intestinal stem cells with dysfunctional telomeres, but this enhancement results in chromosomal instability (Sperka et al. 2012a). Thus, each branch of the p53 checkpoint can compensate for a loss of the other branch, hence maintaining the capacity to deplete genetically instable stem cells (Sperka et al. 2012a). It is possible that chromosomal instability of stem cells contributes to the increased cancer risk that is observed in telomere-dysfunctional mice in the context of p53 deletion (Artandi et al. 2000).
9.5.3 Effects of Altering Upstream Checkpoint Components
Altering the expression of p53 inducers also affects telomere-driven aging. Loss of ATM aggravates DNA damage accumulation and accelerates tissue aging in telomere-dysfunctional mice (Wong et al. 2003). The molecular mechanisms remain to be determined but could involve the role of ATM in oxidative stress responses (Ito et al. 2004; Maryanovich et al. 2012), telomere maintenance (Smilenov et al. 1997) or replication fork stalling (Stiff et al. 2006). The first two processes have already been shown to influence stem cell maintenance, but an effect of replication fork stalling in stem cell aging remains to be investigated.
In contrast to data on ATM, deletion of exonuclease 1 (Exo1) improves stem cell maintenance and tissue aging in telomere-dysfunctional mice (Schaetzlein et al. 2007). Exo1 has 5’–3′ exonuclease activity and is involved in DNA mismatch repair (Wei et al. 2003). Exo1 was first shown in yeast to induce DNA damage signals at dysfunctional telomeres by DNA end resection, which leads to the generation of single-stranded DNA and checkpoint induction via ATR and Chk1 (Maringele and Lydall 2002). Deletion of Exo1 impairs the processing of DNA breaks in mammalian cells with dysfunctional telomeres, which results in decreased generation of single-stranded DNA at breaks and impaired recruitment of the DNA repair factor replication protein A (RPA) (Schaetzlein et al. 2007). In telomere-dysfunctional mice in vivo, Exo1 deletion reduces the processing of DNA damage foci and blunts the induction of ATR and p53 in intestinal stem and progenitor cells. This results in decreased induction of cell cycle arrest and apoptosis, prolonged stem cell and tissue maintenance and lifespan extension (Schaetzlein et al. 2007). Loss of Exo1-dependent DNA damage signalling in telomere-dysfunctional stem and progenitor cells does not lead to an increase in chromosomal instability but impairs the formation of chromosomal fusions (Schaetzlein et al. 2007).
The studies on Exo1 deletion in telomere-dysfunctional mice reveal that Exo1-mediated processing of dysfunctional telomeres contributes to fusion formation, underlining the fatal consequences of fusions for organ maintenance (Rudolph et al. 2001; Schaetzlein et al. 2007). These studies furthermore suggest that naturally shortened telomeres may be processed in different ways dependent on the repair machinery available producing different outcomes for the organism. Intestinal stem cells are able to engage DNA repair machineries involving non-homologous end joining (NHEJ) (Hua et al. 2012). NHEJ had been implicated in telomere processing (Verdun and Karlseder 2007), and deletion of key NHEJ components DNA-PKcs (DNA protein kinase catalytic subunit) or Ku86 caused further lifespan reduction in aging telomere-dysfunctional mice (Wong et al. 2007; Maser et al. 2007; Espejel et al. 2004). DNA-PKcs or Ku86 deletion led to enforced intestinal atrophy due to additional impairments in stem and progenitor cell proliferation in mice with short telomeres compared to mice with wild-type levels of DNA-PKcs or Ku86 (Espejel et al. 2004). Levels of chromosomal fusions were unchanged in the intestinal epithelium of Terc and DNA-PKcs double knockout mice (Maser et al. 2007) indicating that the alternative processing of dysfunctional telomeres in the absence of DNA-PKcs was disadvantageous causing enforced intestinal atrophy.
Deletion of DNA mismatch repair factors, Msh2 or Pms2, both reduced intestinal atrophy in aging telomere-dysfunctional mice (Martinez et al. 2009; Siegl-Cachedenier et al. 2007). Neither telomere length nor signs of chromosomal instability were changed significantly, but checkpoint induction via p53 was reduced. The mechanisms involved are not fully clarified but implicated reduced p21 induction and rescued proliferation in intestinal crypts (Martinez et al. 2009; Siegl-Cachedenier et al. 2007). Interestingly, lifespan in the double knockout mice is not rescued since both knockouts produce an increased tumour load in mice with long telomere reserves, which is not suppressed in late-generation telomerase knockout mice (Martinez et al. 2009; Siegl-Cachedenier et al. 2007).
The examples above demonstrate the altered physiological effects of the DNA damage pathways engaged in the context of dysfunctional telomeres. Careful modulation of those may allow extending organ maintenance without increasing the danger of enhanced tumour formation.
9.6 Cancer
Recently, intestinal stem cells have been identified to be the cells of origin for intestinal adenoma formation (Barker et al. 2007; Sangiorgi and Capecchi 2008




Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
