Fig. 7.1
Scheme of the human haematopoietic hierarchy and relevant cell surface markers. All blood lineage cells are derived from HSC. Differentiation of HSC gives rise to MPP that have lost self-renewal capacity. MPP in turn generate progressively more committed progenitors and eventually terminally differentiated blood cells. Phenotypic cell surface markers are indicated next to each population based on data from (Doulatov et al. 2010 #230; Notta et al. 2011 #231; Majeti et al. 2007 #257; Manz et al. 2002 #289). The yellow box denotes the common phenotype (Lin−CD34+CD38−) shared by HSC and MPP. The pink box denotes the common phenotype (CD34+C38+CD10−CD7−) shared by myeloid progenitors. Abbreviations: HSC haematopoietic stem cells, MPP multipotent progenitors, MLP multilymphoid progenitors, CMP common myeloid progenitors, GMP granulocyte-macrophage progenitors, MEP megakaryocyte-erythrocyte progenitors, ETP early thymic progenitors, NK natural killer. Lin, a collection of surface markers derived from all terminally differentiated populations
A number of in vivo and in vitro experimental approaches have been developed to identify human HSC and to study human haematopoiesis (for reviews, see (Goyama et al. 2015; Doulatov et al. 2012)). These include colony-forming assays in semisolid plates in the presence of a cocktail of cytokines to give colonies of myeloid and erythroid cells, as well as in vitro culture on a variety of murine stromal cell lines capable of supporting the differentiation of primitive human progenitors to the different blood cell lineages (La Motte-Mohs et al. 2005; Baum et al. 1992; Berardi et al. 1997; Miller et al. 1999; Reynaud et al. 2003). In vivo assays, based on the capacity of the stem cells to repopulate the entire haematopoietic system after transplantation into conditioned recipients, have also been developed using a number of animal models. The most widely used in the field have been immunodeficient Rag1-, Rag2-null or Scid mice (for reviews, see (Shultz et al. 2007; Rongvaux et al. 2013)). The xenograft model has been refined by targeting mutation of the IL-2 receptor common gamma chain gene and crossing onto the non-obese diabetic (NOD) background to give mice (NOD-Scid IL2rg−/−, NSG) that have impaired adaptive and innate immunity and so are more efficient in engrafting human haematopoietic progenitors (Traggiai et al. 2004; Pearson et al. 2008; Ito et al. 2002; Shultz et al. 2005). Further improvement of the humanised mice models are being generated by the expression of human cytokines and HLA genes (reviewed in (Brehm et al. 2013)). Overall, these in vivo animal models have been used to test the self-renewal, long-term reconstitution and differentiation potential of human HSCs and have been fundamental to characterise the age-associated changes in HSC phenotype.
Human HSCs have been isolated based on the expression of a number of cell surface markers, the first being the sialomucin protein CD34 (Civin et al. 1984). However, the CD34+ population consists of both multipotent and lineage-committed progenitors, and HSC can be further purified by the exclusion of mature lineage (Lin) and CD38 markers as Lin−CD34+CD38− cells (Bhatia et al. 1997; Hogan et al. 2002). Subsequent studies have shown that human HSCs express the CD90 (Thy1) antigen and identified the stem cells as Lin−CD34+CD38−CD90+CD45RA−, while multipotent progenitors (MPPs) are found in the Lin−CD34+CD38−CD90−CD45RA− fraction ((Doulatov et al. 2010; Majeti et al. 2007) see Fig. 7.1). Human HSCs, like murine HSCs, also have the capacity to efflux vital dyes due to the expression of the P-glycoprotein pump and can be isolated as rhodamine (Rho) dull cells (McKenzie et al. 2007). More recently, using a combination of all these markers, together with the expression of the α6 integrin CD49f, Notta et al. have isolated Lin−CD34+CD38−CD45RA−CD90+RholoCD49f+ cells from human cord blood, which are capable of lymphoid and myeloid repopulation at the single cell level and hence represent the purest population of long-term repopulating human HSCs to date (Notta et al. 2011).
7.3 Age-Associated Changes in the Human HSC Compartment
In humans, advancing age is accompanied by significant changes in the function and composition of mature blood cells. These observations, together with the observed decrease in activity of bone marrow with age and results from HSC transplantation showing that donor age is a strong negative risk factor (Kollman et al. 2001), suggest that the proliferative and regenerative capacity of human HSC diminishes with age. As reviewed in Chap. 6, changes in the HSC compartment with age have been well documented in mice. These age-related changes are primarily characterised by an increase in the number of HSCs but a decline in their function, as indicated by decreased repopulation capacity in competitive transplantation assays, as well as a reduction in the capacity to generate lymphoid lineages. However, differences in HSC aging have been observed between different mouse strains indicating that genetic variation plays an important role in HSC aging (Chen et al. 2000; de Haan and Van Zant 2009), making it difficult to directly extrapolate from these murine studies as to the situation in humans.
Until recently, the effects of age on HSCs in humans have been surprisingly poorly characterised. Early studies were based on indirect observations of committed progenitor or differentiated populations rather than the stem cells themselves (Marley et al. 1999; Gale et al. 1997). More recent studies, including work from our group, have sought to directly assess the frequency and in vitro and in vivo differentiation potential of HSC populations isolated from the bone marrow of young and elderly individuals (Taraldsrud et al. 2009; Beerman et al. 2010; Kuranda et al. 2011; Pang et al. 2011). These studies consistently found an increase in the frequency of immunophenotypic HSCs with age, assessed as either CD34+CD38− or more stringently as CD34+CD38−CD90+CD45RA− cell populations (Table 7.1). They further suggest that MPPs may also increase in elderly bone marrow, although to a lesser extent than primitive HSCs. In contrast, the pool of CD34+ cells, which includes both multipotent and lineage-committed progenitors, remains unchanged (Table 7.1). Functional analysis did not reveal a significant difference in the engraftment efficiency of elderly compared to young CD34+ cells in xenotransplantation experiments in NSG mice (Kuranda et al. 2011). However, since the frequency of CD34+CD38− HSCs is increased in the CD34+ population of the elderly, this suggests that there is a per cell decrease in stem cell repopulating activity with age. This was indeed found to be the case when limiting numbers of more purified CD34+CD38−CD90+CD45RA− HSCs were transplanted in NSG mice (Pang et al. 2011), showing that in humans, as in mice, aging is associated with a decrease in HSC function.
Table 7.1
Summary of age-related changes in the frequency of haematopoietic progenitors in human bone morrow (found in the literature)
Cell type | Phenotype | % MNC | Fold change | BS | References | |
|---|---|---|---|---|---|---|
Young | Old | |||||
HSC | CD34+CD38− | 0.1a | 0.7a | 7.0 ↑ | P < 0.01 | Taraldsrud et al. (2009) |
CD34+CD38− | 0.11 | 0.45 | 4.1↑ | P < 0.005 | Kuranda et al. (2011) | |
Lin-CD34+CD38−CD90+ | 0.04 | 0.1 | 2.5↑ | P < 0.0002 | Beerman et al. (2010) | |
Lin-CD34+CD38−CD90+CD45RA− | 0.08 | 0.25 | 3.1↑ | P < 0.002 | Pang et al. (2011) | |
MPP | Lin-CD34+CD38−CD90−CD45RA− | 0.18 | 0.23 | 1.3 | ns | Pang et al. (2011) |
CD34+CD38−CD10−CD90−CD45RA− | 0.11 | 0.22 | 2.0 | ns | Kuranda et al. (2011) | |
MLP | CD34+CD38−CD10+CD90−CD45RA+ | 0.01 | 0.02 | 2.0↑ | P < 0.005 | Kuranda et al. (2011) |
CLP | Lin-CD34+CD38+CD127+ | 0.08 | 0.04 | 2.0↓ | P < 0.05 | Pang et al. (2011) |
B/NK | CD34+CD38+CD90−CD45RA+CD10+ | 0.44a | 0.08a | 5.5↓ | P < 0.02 | Kuranda et al. (2011) |
B committed | CD34+CD19+ | 0.44a | 0.22a | 2.0↓ | P < 0.01 | Kuranda et al. (2011) |
B cells | CD19+ | 3.2 | 2.5 | 1.3 | ns | Kuranda et al. (2011) |
CMP | Lin-CD34+CD38+CD123+CD45RA− | 0.045 | 0.045 | 1.0 | ns | Pang et al. (2011) |
GMP | Lin-CD34+CD38+CD123+CD45RA+ | 0.04 | 0.04 | 1.0 | ns | Pang et al. (2011) |
MEP | Lin-CD34+CD38+CD123−CD45RA− | 0.04 | 0.038 | 1.1 | ns | Pang et al. (2011) |
Early myeloid | CD34+CD38+CD90−CD45RA+/−CD10− | 0.8a | 0.7a | 1.1 | ns | Kuranda et al. (2011) |
Myeloid committed | CD34+CD33+ | 0.9a | 1.2a | 1.3 | ns | Kuranda et al. (2011) |
CD34+progeniters | CD34+ | −2 | −2 | 1.0 | ns | |
CD34+progeniters | CD34+ | 3.5 | 3.6 | 1.0 | ns | Taraldsrud et al. (2009) |
In addition to decreased repopulating activity, CD34+CD38−CD90+CD45RA− HSCs from elderly individuals appeared to be less efficient at reconstituting lymphoid cells and are more myeloid biased than young HSC in engraftment assays (Pang et al. 2011). Consistent with a loss of lymphoid potential, the frequency of early lymphoid progenitors (CLP and B/NK progenitors) and committed B cell precursors is reduced in human bone marrow with age, while myeloid lineage progenitors (early myeloid, CMP, GMP, MEP) and committed myeloid cells remain stable (Table 7.1). Furthermore, elderly HSCs exhibit a diminished capacity to generate B cells in vitro in differentiation assays on stromal cells (Kuranda et al. 2011; Pang et al. 2011). Age-related changes in the differentiation potential of elderly HSCs are associated with increased expression of genes specifying myeloid fate and function and downregulation of genes involved in lymphopoiesis (Pang et al. 2011). Altogether these studies clearly show that aging in humans is associated with an increase in phenotypic HSCs, but that the stem cells that accumulate with age are functionally deficient, both in terms of repopulating activity and differentiation potential (summarised in Fig. 7.2). In this respect, human HSC aging resembles the C57BL6 mouse model of aging (see Chap. 6).
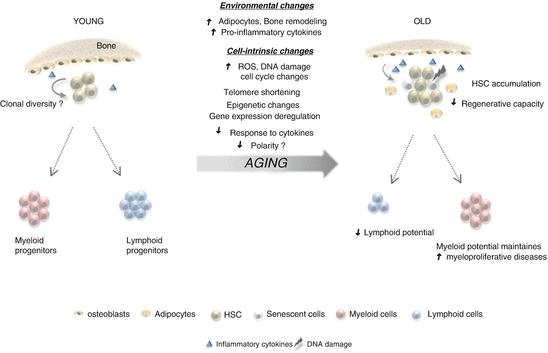
Fig. 7.2
Overview of human HSC aging. Although phenotypic HSC increase with age, both regenerative capacity and lymphoid potential of HSC is diminished. In contrast, myeloid potential is stable or even increased, leading to a propensity towards myeloproliferative disease. Potential mechanisms leading to the decline of HSC functions are indicated above the arrow and include both intrinsic modifications of HSC and environmental changes within the bone marrow milieu. Decreased cell polarity with age has not yet been demonstrated in human HSC
The molecular mechanisms responsible for the changes in human HSC function with age are still being elucidated. In mice it has been demonstrated that HSCs are not homogeneous and that they can be further stratified into subpopulations which have a myeloid-biased or lymphoid-biased/balanced differentiation potential (Muller-Sieburg et al. 2002; Challen et al. 2010; Dykstra et al. 2007). Continuing from this observation, it was shown that the age-associated myeloid skewing in output observed in mice reflects a selective expansion of myeloid-biased stem cells in the aging HSC pool (see Chap. 6). At present, the existence of distinct lineage-biased subpopulations has not been demonstrated directly in human HSCs. It therefore cannot yet be stated conclusively whether the age-dependent increase in myeloid output seen in humans represents a relative shift in distinct clonal HSC subpopulations as in the mouse (the so-called clonal-shift model of HSC aging) or is the result of uniform age-associated changes in all stem cells.
7.4 Molecular Mechanisms of HSC Aging
7.4.1 Accumulation of DNA Damage
DNA damage is recognised as one of the major mechanisms driving age-associated decline in cell function and thereby leading to eventual tissue and organismal failure (reviewed in (Lombard et al. 2005)). Both human and mouse aging are associated with a decline in the efficiency of DNA repair pathways (Gorbunova et al. 2007) and an accumulation of various types of DNA damage in somatic tissues (Sedelnikova et al. 2004, 2008; Wang et al. 2009). A key role for DNA damage in aging is evidenced by the observation that many human premature aging syndromes are associated with defects in the normal mechanisms of genome surveillance and repair (Best 2009). DNA damage within stem cell pools is likely to be particularly damaging to organismal viability, as it has the potential to not only impact upon the size of the stem cell population itself but also upon the function and integrity of progenitor lineages.
Two studies have investigated DNA damage in aging human HSCs by comparing the number of foci of phosphorylated histone H2AX (γH2AX), an early response to DNA double-stranded breaks (DSB), in immunopurified haematopoietic stem populations from humans of different ages (Rube et al. 2011; Yahata et al. 2011). Both studies demonstrated that aging was associated with a significant increase in the frequency of γH2AX foci in CD34+CD38− HSCs. The study by Yahata et al. (2011) demonstrated that this age-associated increase in DSB was correlated with an increase in the intracellular concentration of reactive oxygen species (ROS), suggesting that oxidative stress is an important driver of DNA damage in aging HSCs. Further, they demonstrated that experimentally inducing ROS and DNA damage in HSCs led to a decrease in HSC reconstitution potential. As ROS are a normal cell-intrinsic by-product of oxidative metabolism and cell cycling, it is possible that this increase in ROS and DNA damage reflects an increase in the portion of HSCs that have entered cycle with age. Similarly, increased ROS and DNA damage is also observed when HSCs are forced into cycle by transplantation, and this is also correlated with loss of HSC repopulation capacity (Yahata et al. 2011). Overall, this study demonstrates that DNA damage due to oxidative stress does increase in human HSCs with age and that this damage impacts upon normal HSC function, in terms of reconstitution in engraftment assays. Consistent with these findings, gene expression profiling in human HSCs indicated that aging is associated with the upregulation of genes involved in DNA damage repair, hinting at the possibility that aged HSCs have activated repair pathways (Pang et al. 2011). As discussed above, the number of human HSCs is increased with age, suggesting that the effects of DNA damage are not simply due to cytotoxic and cytostatic depletion of the stem cell pool and are more likely to reflect functional changes. It will be interesting to determine whether the effects of increasing intracellular ROS and DNA damage also induce other aspects of HSC aging, such as changes in differentiation potential and an increased incidence of mutations and chromosomal translocations observed in haematological malignancy.
Surprisingly, several studies have revealed important differences between how mouse and human HSCs respond to DNA damage. In response to low levels of ionising radiation, normally quiescent mouse haematopoietic stem and progenitor cells (Lin−c-Kit+Sca-1+Flk2−) appear relatively resistant to apoptosis and instead survive the damage by undergoing non-homologous end joining, which is an innately error-prone DNA repair pathway (Mohrin et al. 2010). In contrast, human HSCs (Lin−CD34+CD38−) appear to adopt apoptosis as the default programme in response to radiation-induced damage (Milyavsky et al. 2010). Thus, these two studies suggest that humans and mice have adopted different approaches to survive genotoxic damage within the HSC compartment, with mice favouring immediate survival of the stem cell pool, but at the risk to long-term fitness in the form of mutagenic change during repair, and humans favouring removal of the damaged cells. It remains to be seen whether this surprising observation reflects technical differences between these studies or is indicative of an evolutionary response to the different life spans of these two mammals (Lane and Scadden 2010).
7.4.2 Telomere Shortening
As well as DSB, another form of DNA damage that has been characterised with age in human HSCs is telomere shortening. In most human somatic cell types, the telomere loses some 50–200 bp of terminal repeat sequence with every round of cell division as a result of incomplete end replication (reviewed in (Greenwood and Lansdorp 2003)). When telomeres reach a critically short threshold, cells are triggered to either enter a senescent state or to undergo apoptosis (Bodnar et al. 1998). In some cases, and particularly in the germ line, telomeres are protected against shortening by the activity of telomerase (Morin 1989). While telomerase is not expressed in most somatic tissues (Kim et al. 1994), telomerase activity is readily detectable in both human (CD34+CD38−) and mouse (Thy-1.1loSca-1+Lin−Mac-1−CD4−c-kit+) HSCs and haematopoietic progenitors, albeit at low levels (Yui et al. 1998; Morrison et al. 1996; Broccoli et al. 1995). Despite this telomerase competence, telomere shortening has been observed with advancing age in human HSCs and progenitor populations (Vaziri et al. 1994) suggesting that these low levels of telomerase activity are not sufficient to completely protect against telomere shortening in the HSCs of aged individuals. That telomere maintenance is critical for normal HSC function throughout life is suggested by the observation of acute bone marrow failure in dyskeratosis congenita and acquired aplastic anaemia, diseases associated with deficiencies in telomerase core components (reviewed in (Townsley et al. 2014)). Further, telomerase knockout mice exhibit impaired HSC function including a myeloid-lymphoid skewing (Ju et al. 2007) typical of normal age-associated changes in both mouse and human HSCs. However, this skewed phenotype is mainly due to changes extrinsic to the HSC themselves, including effects of telomere shortening on the stem cell niche (Ju et al. 2007) and the systemic environment (Song et al. 2010). Thus, the intrinsic effects of telomere shortening on HSC function are currently unclear.
7.4.3 Cell Cycle Changes and Senescence
HSCs are predominantly a quiescent population. In humans it has been shown that only a small proportion (~10 %) of HSCs are in cycle at any time (Pang et al. 2011; Laurenti et al. 2015). This primarily quiescent state appears important to minimise replication-associated mutations which could otherwise accumulate in this long-lived cell type, and in mice it has been shown that disruption of HSC quiescence leads to the accumulation of DNA damage and eventual stem cell exhaustion (Cheng et al. 2000; Walter et al. 2015). In a recent study, cell cycle profiling of human HSCs (Lin−CD34+CD38−CD90+CD45RA−) isolated from the bone marrows of young (20–35 years) and old (>65 years) individuals revealed that aging was associated with a significant increase in the proportion of cells which had exited the quiescent (G0) state and entered G1 (Pang et al. 2011). Consistent with this observation, this study also found that among the genes upregulated with aging, there was significant enrichment for genes involved in cell cycle progression.
The relationship between quiescence and DNA damage is complex. While ostensibly protecting against replication-driven DNA damage, it appears that this quiescent state may also contribute to the accrual of long-term DNA damage with age, due to the attenuation of checkpoint control and DNA damage response pathways in quiescent cells (Rossi et al. 2007; Beerman et al. 2014). Consistent with this, it appears that the cells emerging from quiescence with age are functionally attenuated and may be undergoing cell cycle arrest in G1 due to the activation of cell cycle checkpoints in response to DNA damage, leading to an eventual senescence-like state. In support of this, the age-associated increase in the number of HSCs in G1 does not translate into expansion of downstream progenitors ((Kuranda et al. 2011; Pang et al. 2011) and Table 7.1). Furthermore, it has been reported that expression of the G1 checkpoint protein p16INK4a increases with age in the HSC compartment and that p16INK4a deletion improves HSC function in old animals (Janzen et al. 2006). It will be interesting in the future to isolate live HSCs at different stages of the cell cycle, so that the properties of this emerging G1 population can be investigated directly at the molecular and functional levels.
7.4.4 Epigenetic Changes
Epigenetics refers to forms of information that regulate gene expression and thereby influence cellular phenotype, but that does not involve modifications of the DNA sequence itself. Epigenetic signals can be extremely plastic, making them ideal for controlling dynamic changes in gene expression profiles that drive cell fate determination, lineage commitment and differentiation. The most important forms of epigenetic information known at present are changes in chromatin structure due to chemical modifications of DNA (DNA methylation and hydroxymethylation), covalent modifications of the histone proteins and remodelling of nucleosomes, as well as the subnuclear localisation of genes. Widespread changes in epigenetic chromatin structure and nuclear organisation have been observed during aging in a number of organisms, including yeast, C. elegans, Drosophila and mammals (reviewed in (O’Sullivan and Karlseder 2012)).
It is now clear from studies in human and mouse that epigenetic signals are critically important in maintaining HSC differentiation plasticity and multipotentiality by priming cells for the expression of different lineage-associated transcription programmes and that this permissive epigenetic programme becomes progressively restricted following lineage choice and commitment (Maes et al. 2008; Weishaupt et al. 2010). Thus, perturbation of the normal epigenetic state of HSCs could potentially have significant effects on phenotypic plasticity and differentiation output, similar to those observed with aging. Indeed, recently published high-throughput epigenomic profiling studies in mice have demonstrated that aging is associated with genome-wide changes in the epigenetic state of mouse HSCs, contributing to gene expression changes that underlie some of the observed functional alterations, including perturbed self-renewal and differentiation potential (Beerman et al. 2013; Taiwo et al. 2013; Sun et al. 2014).
While comparable epigenomic studies are still awaited in humans, emerging lines of evidence suggest that, as in the mouse, epigenetic change is also likely to be an important driver of the functional changes observed with age in human HSCs. Perhaps the epigenetic signal, which has been best characterised during HSC aging in humans, is DNA methylation. DNA methylation is the addition of a methyl group to the pyrimidine ring of cytosine residues, which are usually located within the symmetrical dinucleotide CpG, to form 5-methylcytosine (5-meC). Methylation of genomic DNA is associated with transcriptional repression and is a key regulatory mark in both normal development and in disease (Schubeler 2015). The genome-wide levels of promoter DNA methylation have been compared between human CD34+ haematopoietic progenitors from cord blood and adult bone marrow (Bocker et al. 2011). This study found significant age-associated changes in DNA methylation, which exhibited a bimodal distribution; ~60 % of changed genes showed depleted methylation with age, and ~40 % had gained methylation. Interestingly, there was a significant overlap between genes which became hypomethylated in human HSPC with age and those that become hypomethylated during myeloid differentiation, leading the authors to speculate that these age-associated methylation changes may explain the myeloid-biased output observed in aged HSCs, by allowing the premature activation of the myeloid programme and loss of stem cell plasticity (Bocker et al. 2011). These findings now need to be confirmed in more purified populations of human HSCs and by comparing adult HSCs from different ages, but are highly consistent with the bimodal pattern of DNA methylation changes with age which have been reported in adult mouse HSCs (Taiwo et al. 2013; Sun et al. 2014) as well as the functional changes observed in mice lacking DNA methyltransferases (Challen et al. 2014). Together these studies suggest that DNA methylation could play a key role in maintaining HSC plasticity and suggest that age-associated changes in DNA methylation could underlie important aspects of the aging HSC phenotype.
Covalent histone modifications have been shown to play a key role in stem cell plasticity and lineage choice (Bernstein et al. 2006; Mikkelsen et al. 2007). The study of histone modifications in human HSCs has shown that lymphoid and myeloid loci that are not expressed in HSCs, but that will be expressed in downstream committed progenitors, are marked by high levels of active histone modifications (H3K4me2 and H4ac) in HSCs (Maes et al. 2008




Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
