Fig. 4.1
Cutaneous carcinogenesis of SCC in OTR
4.3 Effects of Individual Immunosuppressive Drugs (Table 4.1)
Table 4.1
Effects of individual immunosuppressive drugs
Drug | Effect | Literature |
|---|---|---|
Glucocorticoids | Possibly impair tumor surveillance, increase risk for SCC, BCC, and non-Hodgkin lymphoma | |
Calcineurin inhibitors | Increase the expression of TGF beta, VEGF (increasing angiogenesis and tumor growth), and ATF3 (impacting p53), decreases DNA repair following UV exposure | |
Increased risk for SCC | ||
Azathioprine | Photosensitizes to UVA and increases DNA damage, reduces DNA repair activity | |
Increases risk for SCC more than BCC | ||
Mycophenolate mofetil | Antioxidative activity by inhibition of 2 NAD(p)H oxidase, reduces angiogenesis | |
Lower risk for NMSC than other antimetabolites, no risk for lymphoma | ||
mTOR inhibitors | Inhibit IL-2 signaling, antitumoral and antiangiogenic potential (inhibits VEGF and TGF beta) | |
Lower risk for SCC than CNI |
4.3.1 Glucocorticoids
Several studies have addressed the association of steroid use with cutaneous carcinogenesis. The use of systemic glucocorticoids for more than 1 month in diverse indications other than OTR doubles the risk for squamous cell carcinoma (relative risk for steroid use 2.31 (95 % confidence interval 1.27–4.18)). Oral glucocorticoids increased the risk for SCC in patients other than OTR (adjusted odds ratio = 2.31; 95 % CI = 1.27, 4.18) [20], while the risk of BCC did not change. A Danish study found an increased risk for basal cell carcinoma, squamous cell carcinoma, and non-Hodgkin lymphoma in patients on systemic glucocorticoids longer than a month in non-OTR patients [21]. Additional studies suggest oral glucocorticoids as a risk factor for slightly increased BCC risk in the general population where the ratio of incidence increased to 1.17 (95 % CI: 1.08–1.28) for use beyond 1 year and to 1.22 (95 % CI: 1.09–1.36) for use beyond 5 years before the development of BCC [22]. On the other hand, inhaled corticosteroids did not have an impact of SCC and BCC formation, probably due to the modest systemic steroid absorption [20].
Until now, a mechanism for glucocorticoids in skin cancer formation has not been defined. It is assumed that glucocorticoids reduce the tumor surveillance by the immune system in multifactorial ways.
4.3.2 Calcineurin Inhibitors
Calcineurin inhibitors (CNIs) such as cyclosporine A (CsA) and the ascomycin-related drugs tacrolimus and pimecrolimus belong to this class. They inhibit calcineurin mainly in lymphocytes, thus impeding the nuclear localization of the transcription factor nuclear factor of activated T cells (NFAT). This blockade represses cytokines and chemokines needed for T-cell activation. The result is a profound systemic suppression of the immune system focused on T lymphocytes (reviewed in Reynolds and Al-Daraji [19]).
In OTR with systemic calcineurin inhibitor therapy, there is an immediate rise in incidence of SCC directly after transplantation without delay. Stopping CNI shows a decrease of the SCC incidence [23]. Compared to standard CsA dose (150–250 ng/m), a low-dose CsA therapy (75–125 ng/mL) shows a decrease of NMSC incidence. In the standard regime, 26 patients out of 115 suffered of NMSC compared with 17 out of 116 patients in the low-dose group. Unfortunately, patients with lower CsA therapy demonstrated more transplant rejection episodes as a limiting problem [24]. A single-center cohort study compared the outcome of cancer including skin cancer following renal transplantation from CsA in 798 patients to tacrolimus in 355 patients. The study concluded that there was no difference between two groups regarding skin cancer formation for the use of induction or maintenance therapy [25].
CNI leads to cancer formation by increasing the expression of transforming growth factor beta (TGF beta) [26]. A dose dependency was seen for tacrolimus on tumor growth and TGF-beta1 cytokine expression in mice, where this overexpression by tacrolimus provides immunosuppressive signals and contributes to tumor progression [27]. Compared to CsA, tacrolimus is associated with a minor increase of the TGF-b transcription rate in humans [28]. CsA also induces the expression of vascular endothelial growth factor (VEGF) [29]. Neoangiogenesis driven by VEGF probably drives tumor growth and metastasis. While general immunosuppression by CNI is certainly driving skin cancer formation, CNI also directly affects keratinocytes and thus contributes to cancer formation of the skin in particular. Several mechanisms have been delineated as contributors to the clearly recognizable increase in tumor formation for patients on these compounds.
DNA repair was shown to be inhibited in lymphocytes from OTR on CsA and ascomycin, lagging behind in compensating for UV-induced damage. The UV-induced apoptosis in human keratinocytes was impaired as well. Both of these phenomena were observed at therapeutic doses of CsA. Apoptosis and necrosis were less visible at low CsA doses of 125 and 250 nM, with a more pronounced effect at 125 nM [30]. The persisting DNA damage is a risk factor for skin cancer formation. Apoptosis protects our cells from cancer formation including skin cancer, whereas the reduction of apoptosis by CsA is an initiating factor for cancer. In summary, CNI increases NMSC formation in two ways: DNA damage and the reduction of apoptotic removal of such cells [31].
CsA shows effects on the mitochondria where it inhibits the opening of mitochondrial permeability transition pore (MPTP). Oxidative stress induced by UV light leads to opening of MPTP, mediating cell death. CsA now promotes survival of damaged cells, allowing for tumor development. The model used showed CsA to prevent cell death in this manner, while TAC had no such effect. CsA rather than TAC may thus drive cutaneous carcinogenesis by prolonged keratinocyte survival following genotoxic stress [30].
Xunwei Wu et al. [32] reported that under CNI, the transcription factor ATF3 is selectively upregulated. ATF3 belongs to the family of AP-1 transcription factors in keratinocytes. ATF3 has a direct impact of tumor formation in keratinocytes by downregulating the mRNA expression of p53 and consecutively the p53-dependent senescence. Recent data demonstrated that UVA light increases ATF3 expression in keratinocytes also in the general population independent of CNI medication. This may be the reason why SCC appears in OTR on CNI preferably on chronically sun-exposed skin [33, 34].
In summary, CNI increases skin cancer formation both by immune-mediated effects such as reduced tumor surveillance by inactivation of lymphocytes and TGF beta secretion, promotes growth and metastasis by VEGF, and directly drives keratinocytes into skin cancer by reduced DNA repair following UV damage, interference with mitochondrial MPTP, and increased ATF3 expression impacting p53 which is potentiated by UVA.
4.3.3 Azathioprine
Azathioprine (AZA) is a purine analog which inhibits purine synthesis and metabolism by incorporation of its metabolite 6-thioguanine into the DNA. OTRs with long-term use of azathioprine have an 8.8-fold increased risk of nonmelanoma skin cancer [35]. Immunosuppressive therapy in organ transplant recipients has a stronger impact on the incidence of squamous cell carcinoma than on basal cell carcinoma [13]. AZA counts as risk factor for developing SCC after a total treatment duration of more than 11 years or a cumulative individual dose of more than 500 g AZA. Patients being on AZA therapy for autoimmune inflammatory rheumatism were recognized to have a significantly increased risk of developing SCC (OR 30 (95 % CI 2.6–345.1) and OR 13.5 (95 % CI 1.3–143.6)) without correcting for risk factors, compared to the general population. After the start of AZA, the time to develop SCC ranged from 3.5 to 15.2 years (median 11.3 years) [36].
AZA not only leads to cancer formation by immunosuppression, it is – not unlike CNI – also directly carcinogenic. AZA increases the oxidative DNA damage caused by UVA irradiation in keratinocytes [37]. UVA damages DNA by radical oxygen species in keratinocytes. AZA converts to its active metabolite 6-thioguanine (6-TG) inserted as base analog into DNA of dividing cells. 6-TG DNA preferentially absorbs UVA and, as a first step in cancer formation, induces DNA strand breaks [37]. Kidney transplant recipients under AZA treatment showed a reduced minimal erythema dose for UVA and an increase in p53 mutant foci, while the DNA repair activity was reduced in human keratinocytes [38]. In hairless albino mice, AZA increases the risk of ultraviolet radiation-induced skin cancer. Thirty weeks after the start of UVA and AZA treatment, the mean number of cancers per mouse was 4.38 (p < 0.05) [39]. Stopping azathioprine normalizes the photosensitivity to UVA, while this process takes up to 2 years [40].
4.4 Mycophenolate Mofetil
Mycophenolate mofetil (MMF) is the ethyl ester of the fungal antibiotic mycophenolic acid, which inhibits the de novo purine biosynthesis pathway. MMF is currently widely used in replacement of AZA and is part of a typical initial immunosuppressive regimen in kidney transplantation.
Compared with other antimetabolites, MMF was proven to be a lower risk for malignancies in general, including skin cancer [41]. James et al. looked for associations in the development of malignancy [42]. Patients on MMF therapy had a lower incidence of skin cancers compared to patients on AZA (adjusted RR = 0.73, 95 % confidence interval 0.25–1.03, p = 0.09). There was also a slightly lower risk for PTLD/lymphoma in the MMF group (adjusted RR = 0.44, 95 % confidence interval 0.19–1.01, p = 0.054). In total, the adjusted risk for skin cancer formation was reduced on MMF by 27 % (p value 0.02, relative risk 0.73, 95 % confidence limits for relative risk 0.56–0.95) [42]. Another positive finding was the unchanged risk for lymphoma formation on MMF compared to patients receiving other immunosuppressive treatments [43].
MMF is known to reduce angiogenesis and consequently tumor growth. Studies proclaim antioxidant activity for MMF. In contrast to calcineurin inhibitors like CsA, MMF decreases endothelial O2 formation (superoxide anions) by inhibition of the 2 NAD(P)H oxidase. CsA increased the 2 NAD(P)H oxidase activity resulting in O2 formation. Endothelial dysfunction is induced by increases in production of reactive oxygen species, such as superoxide anions produced by NAD(P)H oxidase [44].
4.4.1 Mammalian Target of Rapamycin (mTOR) Inhibitors
Inhibitors of the mammalian target of rapamycin (mTOR), including sirolimus and everolimus, are newer immunosuppressants. mTOR participates in key cellular processes such as protein translation, ribosome biosynthesis, and the regulation of the cell cycle. The immune system is influenced by mTOR at the level of lymphocyte proliferation, as well as angiogenesis and wound healing, and the proliferation of cancer cells [45].
In T cells, mTOR inhibitors block IL-2 signaling: sirolimus – a structural analog of the macrolide antibiotic FK 506 – enters into a complex with the intracellular FK-binding protein-12 (FKBP12) which exhibits high affinity to mTOR. mTOR inhibition dephosphorylates and inactivates in turn the p70 ribosomal protein S6 kinase. In consequence, the cell cycle is arrested at the juncture of G1 and S phase. As a result, IL-2 activation of lymphocyte proliferation and immunoglobulin production are impaired [46].
Sirolimus is a potent non-nephrotoxic immunosuppressant which – similar to MMF – has been recognized to reduce the incidence of malignancies in OTRs by its antitumoral and antiangiogenic features [47, 48].
The effect of sirolimus on the growth of various cancer cell lines has been studied in mice. Compared with control mice, sirolimus-treated mice have shown a diminished tumor growth. In sirolimus-treated mice, VEGF secretion and subsequently the circulating levels of VEGF and TGF-beta1 were lower [2, 45].
VEGF is a key mediator of angiogenesis. mTOR inhibition by sirolimus mediates antiangiogenic effects by lowering VEGF and reducing the endothelial response to VEGF. In addition to the antiangiogenic action, sirolimus inhibits proliferation of tumors in mice [29].
There are two separate mTOR-signaling complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Sirolimus inhibits the mTOR pathway by directly binding mTORC1, whereas everolimus, a derivative of sirolimus, targets the mTORC1 protein, not the mTORC2 protein. Sirolimus and everolimus share a common structure and mechanism but differ in their serum half-life (60 h for sirolimus and 30 h for everolimus, respectively) [3]. Compared to AZA and CsA, mTOR inhibitors have a protective antitumoral effect including for NMSC in OTRs by controlling signals of the carcinogenic pathway and are still powerful immunosuppressive agents [45].
In comparison to conventional immunosuppressive agents, it is believed that mTOR inhibitors also show antitumoral effects through their influence on immune cells. Six months after switching from CNI and AZA to sirolimus FOXP3+ T cells and NK cells, numbers are increased. It is unclear whether discontinuance of CNI/AZA or the introduction of mTOR inhibitors is responsible for these changes in immune phenotype. Elevation of NK cell levels should act protectively against NMSC formation, but the elevation of FOXP3+ T cell number is believed to carry the risk of later cancer formation. The increase of FOXP3 T-cell levels in OTRs after discontinuance of CNI and/or switching to mTOR inhibitors could be a marker for transplant recipients who are at higher risk of new NMSC formation even with the antitumoral impact of sirolimus [49].
Mathew et al. compared in 1,295 kidney transplant recipients different sirolimus therapy regimes and the outcome after 2 years post transplantation regarding malignancies in general. The first group received sirolimus in continuous combination with CsA, the second, sirolimus as base therapy, and the third, sirolimus maintenance therapy after early withdrawal of CsA. The results of the first group demonstrated that 2 years after transplantation, patients receiving placebo had a higher (6.9 %) incidence of malignancies than OTRs receiving sirolimus in continuous combination with CsA. In the second group, kidney transplant recipients receiving sirolimus as base therapy had no malignancies compared with a 5 % incidence in patients receiving CsA. The malignancy rate in the third group was higher in OTRs receiving sirolimus plus CsA compared to patients on sirolimus therapy with early elimination of CsA (Fig. 4.2) [50].
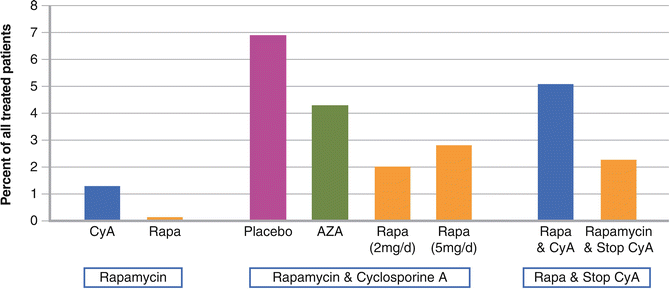
Fig. 4.2
Skin cancer incidence in registration studies for rapamycin. Skin cancer was not a primary objective in these studies but assessed as a secondary parameter
Some registration studies for sirolimus reported as secondary outcome parameters a lower skin cancer incidence in OTR on sirolimus compared to those on calcineurin inhibitors (with either first-time therapy [50] or after switching [51–53]).
De Fijter [55] examined the use of the mTOR inhibitors in 53 renal transplant recipients developing skin cancer post transplantation. In 37 patients, epithelial skin cancer regressed. The tolerability of both sirolimus and everolimus seems to be good with minimal adverse events, pointing to mTOR inhibitors as a tool in the management of epithelial skin cancer in OTR [47, 48, 54, 55]. Existing SCCs post transplantation demonstrated a reduction of thickness and vascularization after the switch from CNI to sirolimus [56].
The CONVERT trial examined 555 renal allograft recipients who converted to a CNI-free and sirolimus-based therapy. Two years after the switch from CNI to sirolimus, the conversion group suffered fewer malignancies in general, including NMSC, compared to those who continued on CNIs (2.2 % vs. 7.7 %; p < 0.001). The graft and patient survival were equal in both groups [52].
A 2012 published study with 86 kidney transplant recipients was about in line with the CONVERT trial. The NMSC rate was reduced with sirolimus (1.31 vs. 2.48 lesions/patient-year; p = 0.022). Squamous cell carcinoma occurred at a lower rate in the sirolimus versus CNI group (p = 0.038), while the rate of basal cell carcinoma was indifferent. A lower percentage of OTR on sirolimus showed new or recurrent NMSC (56.4 % vs. 80.9 %; p = 0.015) or new squamous cell carcinoma (41.0 % vs. 70.2 %; p = 0.006) [51].
The TUMORAPA study observed kidney transplant recipients on CNI therapy with at least one cutaneous squamous cell carcinoma. One group was converted to sirolimus, while the other continued CNI treatment. Converting the immunosuppression from CNI to sirolimus resulted in a reduced risk for subsequent skin cancers, with disease-free survival longer for OTR on sirolimus than on CNI. In summary, 14 patients (22 %) on sirolimus developed new squamous cell carcinomas (6 after stopping sirolimus), while 22 (39 %) of OTR on CNI did so (median time until onset, 15 vs. 7 months; P = 0.02). The relative risk for SCC in OTR on sirolimus stood at 0.56 (95 % confidence interval, 0.32–0.98). Kidney graft function did not change in the either study group. Switching from CNI to sirolimus showed therefore an antitumoral effect among transplant recipients with previous squamous cell carcinoma [53].
The data suggest that the earlier the conversion occurs after an initial diagnosis of cutaneous squamous cell carcinoma, the greater the efficacy. The effect was clear for those OTR with a first SCC (hazard ratio, 0.03; 95 % CI, 0.0–0.91) but not for those with more than one SCC (hazard ratio, 0.67; 95 % CI, 0.29–1.54) [53].
A small German study investigated if mTOR inhibitors could also be a safe immunosuppressive strategy for renal transplant recipients without having had a previous skin cancer. The study included all forms of histologically confirmed nonmelanoma skin cancer and premalignancies. One year after conversion, NMSC was reported in one out of 16 patients in the sirolimus conversion group compared with eight out of 17 in the control group (p
 Update on Staging, Definition, and Chemoprevention of “High-Risk Squamous Cell Carcinoma” in Organ Transplant Recipients
Update on Benign and Inflammatory Skin Disease Secondary to Transplant Medication
Update in Melanoma in Organ Transplant Patients
Update on Staging, Definition, and Chemoprevention of “High-Risk Squamous Cell Carcinoma” in Organ Transplant Recipients
Update on Benign and Inflammatory Skin Disease Secondary to Transplant Medication
Update in Melanoma in Organ Transplant Patients
 Advances in Photoprotection
Advances in Photoprotection
 Advances in Management of “High-Risk Squamous Cell Carcinoma” in Organ Transplant Recipients
Advances in Management of “High-Risk Squamous Cell Carcinoma” in Organ Transplant Recipients
 Update on Our Understanding of HPV as a Risk Factor for Cutaneous Squamous Cell Carcinoma in Organ Transplant Recipients
Update on Our Understanding of HPV as a Risk Factor for Cutaneous Squamous Cell Carcinoma in Organ Transplant Recipients




Related posts:
Update on Staging, Definition, and Chemoprevention of “High-Risk Squamous Cell Carcinoma” in Organ Transplant Recipients
Update on Benign and Inflammatory Skin Disease Secondary to Transplant Medication
Update in Melanoma in Organ Transplant Patients
Advances in Photoprotection
Advances in Management of “High-Risk Squamous Cell Carcinoma” in Organ Transplant Recipients
Update on Our Understanding of HPV as a Risk Factor for Cutaneous Squamous Cell Carcinoma in Organ Transplant Recipients
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
