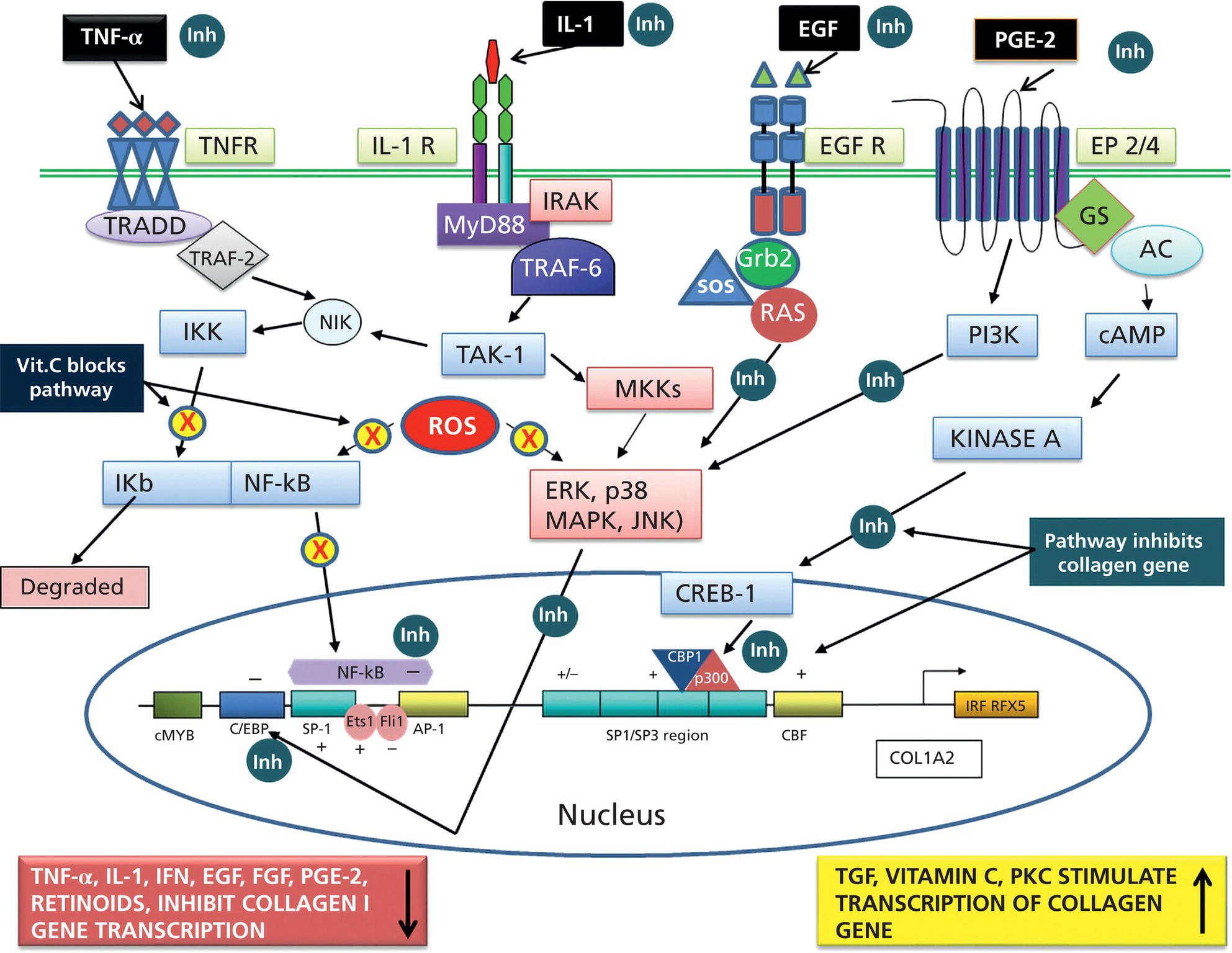
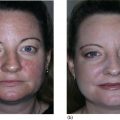
Bryan B. Fuller DermaMedics LLC, Oklahoma City, OK, USA The use of oral and topical antioxidants to prevent skin aging and to provide protection from, and treatments for various diseases, including cancer, has gained considerable popularity over the past 25 years. While it is clear that antioxidants such as vitamins C, E, and A, and carotenoids, can protect cells from free radical damage, it is not clear whether or not taking large quantities of antioxidants can prevent the occurrence of disease or slow the aging process. Some clinical studies have suggested such a role for antioxidants, while other studies have not provided clear evidence that antioxidant supplements can reduce the risk of cancer, heart disease, or aging [1–3]. Regardless of clinical study results, the fact that antioxidants can block free radical damage to cells has led to rapidly growing markets for antioxidant nutritional supplements, antioxidant beverages, and has fueled an entire industry focused on finding foods with ever higher antioxidant potential. Most, if not all, topical skin care products manufactured today contain antioxidants such as vitamin E, vitamin C, and carotenoids, to mention a few. The question of whether or not the antioxidants in topical skin care products, as well as those taken orally, may provide any skin benefits will be explored in this section. An antioxidant is simply a molecule that can prevent the oxidation of other molecules. Their importance in protecting cells from damage results from their ability to block the progression of oxidative damage caused by free radicals. Free radicals are molecules or atoms with unpaired electrons. Having an unpaired electron leaves an incomplete electron shell and makes the atom or molecule more chemically reactive than those with complete electron shells. Because atoms seek to reach a state of maximum stability, they will try to fill the outer shell by “stealing” an electron from another molecule. When the target molecule loses an electron to the free radical, it then, in turn, becomes a free radical and must find a “donor” it can steal an electron from. Thus, a chain reaction begins that causes considerable damage to cellular proteins, lipids, membranes and, DNA. Most free radicals in biological systems are derivatives of oxygen. The most common oxygen radicals in the body are the superoxide anion (O˙2 −) and the hydroxyl radical (˙OH−). While free radicals have extremely short half‐lives, in the order of nanoseconds or microseconds, this is sufficient time to attack molecules and generate new free radicals. In addition to the above oxygen radicals, there are other reactive oxygen species (ROS) which are not actually radicals, but which are reactive and which can cause extensive damage to cells and tissues. Two of the more important of these are hydrogen peroxide and hypochlorite ion [4]. Free radicals and other ROS can cause irreversible and destructive changes to proteins, DNA, and lipids and these can have serious consequences on cell survival, malignant transformation, and the development of disease. One of the most damaging free radical events is lipid peroxidation to membrane lipids which play a key role in cell signaling. In this case, a hydroxyl radical may remove a hydrogen atom from the side chain of a fatty acid, thereby converting the fatty acid into a radical. The fatty acid then reacts with oxygen to form a very reactive peroxyl radical. A chain reaction begins in which one lipid radical becomes two lipid radicals. Ultimately, the lipid radicals may form covalent crosslinks with each other, thereby ending the chain reaction. Unfortunately, the result is crosslinked and functionally damaged lipids. A good example of this is the free radical damage to low‐density lipoprotein (LDL). This damage has been shown to lead to atherosclerosis. Free radicals and other ROS can not only damage proteins, lipids, and other cellular constituents, they can activate signaling pathways that lead to the increased expression of genes controlling inflammation and proliferation. Although ROS are produced constantly by normal cellular events such as respiration, or as a result of enzyme activity, e.g. NADH oxidases, they are continually being inactivated by cellular antioxidants such as superoxide dismutase (SOD), catalase, glutathione (GSH) peroxidase, and GSH. However, if the “load” of ROS in a cell becomes very high due to exposure to such “insults” as ultraviolet radiation (UVR), ionizing radiation, chemotherapeutic drugs, or environmental factors, the endogenous antioxidant mechanisms are overwhelmed, “oxidative stress” occurs, and signaling pathways are activated. Many studies have shown that ROS can activate members of the mitogen‐activated protein kinases (MAPK) family, which includes the growth factor‐regulated extracellular signal‐related kinases (ERKs), the stress‐activated MAPKs, c‐jun NH2‐terminal kinases (JNKs), and p38 MAPKs [5]. Activation of these kinases leads to the enhanced expression of a number of genes including those involved in inflammation and proliferation [6]. In addition, ROS can activate the Nf‐kB pathway further increasing the transcription of genes coding for inflammatory cytokines, as well as for proteins involved in damage to the extracellular matrix (ECM), particularly MMP‐1 [7, 8]. How ROS activate the MAPK and NfkB pathways is not completely understood, but evidence suggests that these free radicals may function by: Regardless of the mechanism of activation, the end result of stimulating the MAPK and Nf‐kB pathways is that gene expression is altered leading to: (1) increased cellular proliferation; (2) increased inflammatory cytokine and chemokine expression; (3) decreased synthesis of collagen; (4) increased COX‐2 expression, resulting in high levels of PGE‐2; (5) cell transformation/carcinogenesis; and (6) increased production of MMPs (matrix metalloproteinases), including MMP‐1, MMP‐2, and MMP‐9, all of which can cause damage to the skin’s matrix [8, 19]. A cartoon depicting some of the inflammatory signaling pathways activated by ROS is shown in Figure 37.1. A large body of recent work has shown a strong link between AGEs (Advanced Glycation End products) and both skin aging and skin inflammation. Aging effects on skin that result from glycation include alterations in skin thickness, texture, resiliency, loss of matrix components, and a decrease in elasticity. Glycation is a non‐enzymatic linkage of sugar to proteins, lipids, and nucleic acids. Specifically, the carbonyl group of simple sugars such as glucose and fructose binds to the amino group of amino acids (especially, lysine and arginine). This initial and reversible reaction forms a Schiff base which is chemically unstable. This Schiff base then re‐arranges to form a more stable ketoamine. Like the Schiff base, this intermediate is also reversible, but because it is reactive, the ketoamine can form irreversible complexes with peptides and proteins, such as collagen, resulting in the production of crosslinks which are non‐reversible and, unfortunately, are resistant to even enzymatic degradation. These large complexes can also undergo additional reactions such as oxidation, polymerization, and dehydration resulting in new forms of AGEs. Since AGEs are resistant to turnover, they accumulate with age, and the skin structure becomes increasingly damaged. ECM proteins are known to be a major target for glycation. Type I collagen is subject to glycation as is elastin and fibronectin. Collagen is not only a key structural component in the dermis, providing mechanical support for tissues, but it also plays an active role in regulating such cell functions as migration, proliferation, and gene expression. Glycation of collagen causes crosslinks between molecules and thus, creates stiffness that reduces skin elasticity. In addition, glycation alters the structure of collagen to the point that it is no longer able to bind correctly to target cells. This leads to lost cell communication. Table 37.1 shows a few of the “aging” effects of glycation on skin cells [20–22]. Table 37.1 Glycation‐mediated skin aging effects. Figure 37.1 Inflammatory signaling pathways activated by ROS and by RAGE. Free radicals produced by UVR can accelerate the development of AGEs. Further, AGEs can trigger the activation of NADPH oxidase and the production of more ROS, which then causes more damage to proteins and lipids and even more AGE formation [23]. Finally, AGEs can activate signaling pathways that cause inflammation [24]. Recent studies have identified a membrane receptor for AGEs, appropriately named Receptor for Advance Glycation End products (RAGE) [25]. Upon binding to AGEs, the RAGE receptor is activated and this activation leads to, not only the activation of NADPH oxidase, but also to the stimulation of several signaling pathway components including MAPKs, ERK 1 and 2, phosphatidyl‐inositol 3 kinase, p21Ras, stress‐activated protein kinase/c‐Jun‐N‐terminal kinase, the janus kinases, and iKK [26, 27]. Stimulating these pathways results in activation of various transcription factors, and, in particular, the activation of nuclear factors kappa‐B (NF‐κB), and AP‐1. These transcription factors activate a variety of matrix‐damaging genes, such as MMP‐1 and MMP‐9, as well as inflammatory genes, including TNF‐α, IL‐1, IL‐8, and COX‐2 [23, 28]. A cartoon showing some of the effects of AGEs binding to its RAGE receptor on the production of inflammatory cytokines is shown in Figure 37.1. Antioxidants protect cells from free radical/ROS damage by either donating an electron to a free radical, thereby stabilizing it and halting the chain reaction, or by accepting the one unpaired electron, again stabilizing the free radical and preventing it from interacting with and damaging proteins, DNA, and lipids. By donating an electron to the free radical to stop the chain reaction, the antioxidant itself becomes a free radical. However, because of its structure, the antioxidant is far less reactive than other radicals. If the antioxidant is relatively large, the effect of the unpaired electron is “diluted” along its structure. The antioxidant “radical” may also be neutralized by another antioxidant or it may be enzymatically restored to its non‐free radical form. GSH is one antioxidant that can donate a hydrogen atom to a hydroxyl radical, thereby neutralizing it. The oxidized GSH is converted back to its reduced form by GSH reductase and is then ready to reduce additional free radicals. Recent studies have shown that antioxidants can prevent free radical damage to the skin in several ways: An antioxidant’s “potency” is based on its capacity to inactivate free radicals, and this is typically quantified by use of ORAC assays. ORAC stands for oxygen radical absorbance capacity and is a method of measuring antioxidant capacities of different foods, vitamins, and compounds [32]. The assay measures the oxidative destruction of a fluorescent molecule (for example, fluorescein) after it is mixed with a free radical producer that generates peroxyl and hydroxyl radicals. By comparing the change in fluorescence in a reaction tube that contains only the fluorescent molecule and free radical with the change in fluorescence measured in the assay tube containing the antioxidant along with the free radical, a measure of antioxidant potential (ORAC score) can be obtained. Table 37.2 shows the ORAC scores of some representative antioxidants. Table 37.2 ORAC values of several antioxidants. Of the many antioxidants that are now being formulated into topical products, two of the most common are vitamin C (ascorbic acid) and vitamin E. Vitamin C is the most prominent antioxidant in the aqueous compartment of cells. It can neutralize a variety of ROS, including hydroxyl, alkoxyl, and peroxyl radicals by hydrogen donation and is thus, an extremely important antioxidant for biological systems. In addition, vitamin C can regenerate oxidized vitamin E and is, in turn, regenerated by GSH. Vitamin C is a required for collagen maturation as it is a necessary cofactor for lysine and proline hydroxylase activity [33, 34]. A deficiency in vitamin C results in a loss of collagen and the development of scurvy [35]. In addition, not only does vitamin C function in the maturation of collagen, but it also stimulates the collagen I and III genes in dermal fibroblasts [36, 37]. Further, by inactivating ROS, vitamin C can also inhibit UVR‐induced levels of inflammatory mediators including PGE‐2, MMP‐1, and TNF‐α [38, 39]. Finally, this vitamin may help in the treatment of cancer [40]. Vitamin C is a useful antioxidant to incorporate into topical products, and, in particular, into sun care products because of its ability to neutralize free radicals generated by UVA and UVB radiation. Unfortunately, vitamin C needs to be continually replaced since even a low dose of UV radiation from the sun can rapidly deplete 30–40% of the vitamin C present in the skin [41]. Vitamin E is one of nature’s most important lipid antioxidants since it associates with membranes and protects the lipid environment by scavenging lipid peroxyl radicals. Vitamin E is found in the stratum corneum where it can act as a “front line” defense against UV radiation‐induced free radicals. By inactivating free radicals, vitamin E can partially protect skin from UVR‐induced erythema, likely by reducing the production of PGE‐2 [42]. As mentioned above, the vitamin E radical can be regenerated by vitamin C, and also by GSH and ubiquinol (coenzyme Q10). While both vitamin C and E are extremely useful antioxidants for protecting the skin from free radical damage, unlike vitamin E and other antioxidants that only inactivate free radicals, vitamin C is a bi‐functional antioxidant in that it has an additional activity that helps prevent damage to the skin caused by inflammatory events. There are many “bi‐functional” antioxidants, like vitamin C, that not only inactivate free radicals, but, in addition, also directly block inflammatory signaling pathways activated by non‐ROS‐mediated events. Because of their ability to reduce inflammation, natural antioxidants are now widely used in both topical and oral products, and considerable research is being directed toward identifying new and more potent natural antioxidants that can be used to address skin inflammation. Inflammatory skin disorders are common and range from occasional rashes accompanied by skin itching and redness to more serious conditions such as atopic dermatitis, rosacea, seborrheic dermatitis, and psoriasis. Skin inflammation is characterized by redness, swelling, heat, itching, and pain, and can exist in either an acute or chronic form with acute disease frequently progressing to a more chronic condition. Acute inflammation can result from exposure to UVR, ionizing radiation, allergens, or to contact with chemical irritants (soaps, hair dyes, etc.). Assuming that the triggering stimulus is eliminated, this type of inflammation is typically resolved within 1 to 2 weeks with little accompanying tissue destruction. A chronic inflammatory condition, however, can last a lifetime, and cause considerable damage to the skin. Some of the cellular and biochemical events which occur in the skin in response to triggering stimuli (e.g. UVR, chemical, or antigen) and which lead to an inflammatory response are shown in Figure 37.2. Figure 37.2 Skin inflammation pathway. Within minutes of exposure of skin to an insult, there is a rapid release of inflammatory mediators from keratinocytes and fibroblasts and from afferent neurons. Keratinocytes produce a number of cytokines and chemokines including PGE‐2, TNF‐α, IL‐1, IL‐6, and IL‐8. Dermal fibroblasts also respond to the insult and to IL‐1 produced by keratinocytes by increasing production and secretion of cytokines including IL‐1, IL‐6, IL‐8 as well as PGE‐2. PGE‐2 increases vasodilation and vascular permeability, facilitates the degranulation of mast cells, and increases the sensitivity of afferent neuronal endings. Increased vasodilation and vascular permeability by PGE‐2 and histamine lead to increased blood flow and extravasation of fluid from blood vessels, resulting in visible redness and swelling. Increased production of TNF‐α and IL‐1 leads to the expression of intracellular adhesion molecules, such as vascular cell adhesion molecule (VCAM) and intercellular adhesion molecule ICAM, on endothelial cells of the blood vessels [43–45]. These proteins, as well as P and E selectin, serve as anchoring elements for blood‐borne monocytes and neutrophils. The attachment of leukocytes to the adhesion molecules slows their movement through the bloodstream and finally causes their adhesion to the endothelial wall. In the presence of chemokines, particularly, IL‐8 produced and released by both keratinocytes and fibroblasts, the adherent leukocytes undergo chemotaxis and migrate from the blood vessel out into the skin where they act to scavenge the area of debris and also produce additional inflammatory mediators. The initial acute response occurs within minutes of the insult to the skin. The subsequent movement of neutrophils and monocytes into the “wounded” area typically takes up to 48 hours to occur. If the triggering stimulus is eliminated, inflammatory mediator production by keratinocytes, fibroblasts, and mast cells ceases, the influx of leukocytes to the “wounded” area decreases and inflammation subsides [46–48]. In contrast to acute inflammation which typically resolves in 1 to 2 weeks, chronic inflammation results from a sustained immune cell‐mediated inflammatory response within the skin itself and is long‐lasting. This response involves antigen presenting cells (APC) in the skin, called Langerhans cells in the epidermis and dendritic cells (DC) in the dermis, that, when activated, transport antigens through the lymphatics and to T lymphocytes where the antigen is “presented”. The T‐lymphocytes are activated and travel back to the skin where they proliferate and express a wide range of inflammatory mediators as well as matrix‐eroding enzymes (MMP‐1; collagenase)[49]. Cytokines produced by T‐lymphocytes can stimulate fibroblasts and keratinocytes to produce additional cytokines and chemokines, and can also induce the expression of a variety of tissue‐destructive enzymes by fibroblasts, including MMP‐1 (collagen), MMP‐3 (stomelysin‐1), and MMP‐9 (gelatinase B). As long as the antigen or insult stimulus persists in the skin, the inflammatory response will continue, resulting in significant and serious tissue destruction. Inflammatory processes in the skin, particularly those triggered by long‐term exposure to solar radiation, trigger the production of ROS and activate molecular pathways that escalate the aging process. Even natural chronological skin aging is due, in part, to increased ROS activity [50]. Actinic aging, or photoaging, that occurs following prolonged exposure of the skin to UV light from the sun results in increased cytokine production with attendant activation of genes in both keratinocytes and fibroblasts that cause erosion of the normal skin structure. MMPs, which break down the skin ECM causing sagging and wrinkling, are stimulated in sun‐exposed skin. Furthermore, dermal fibroblast synthesis and assembly of collagen, which is required to maintain and restore the ECM, is inhibited while elastin production is over‐stimulated, leading to elastosis. It is now widely accepted that sun‐exposed skin in most individuals remains in a constant state of low‐level UV‐induced inflammation, and that this “smoldering” inflammation (often called “inflammaging”) is responsible for the signs of skin aging that appear in middle age [51–54]. As discussed previously, many of the inflammatory events described above are triggered by environmental events that lead to excess ROS production. The ROS then activates signaling pathways in cells that result in the activation of genes involved in skin inflammation. All antioxidants inactivate free radicals and in doing so indirectly prevent the activation of inflammatory signaling pathways by ROS. If, however, inflammation is triggered by an event that activates a signaling pathway that does not involve a ROS, many antioxidants, including vitamin E, cannot block the resulting inflammatory response, and skin damage can result. On the other hand, “bi‐functional” antioxidants can not only inactivate ROS, but can also directly inhibit non‐ROS‐mediated inflammatory signaling pathways. One interesting example of a bi‐functional antioxidant that has these two activities is vitamin C. As mentioned, this antioxidant is a required cofactor for two enzymes that are involved in collagen maturation. In addition, vitamin C stimulates collagen I and III gene transcription. Finally, vitamin C helps maintain normal collagen levels in dermal fibroblasts by two additional mechanisms: (1) the inactivation of ROS produced during the inflammatory response, and (2) the inhibition of cytokine‐activated signaling pathways that repress collagen gene activity. The regulation of the collagen I gene is fairly complex with the upstream promoter region having a number of regulatory sites that respond, either positively or negatively, to transcription factors activated by signaling pathways. A cartoon showing some of the regulatory sites on the collagen I gene promoter, as well as the complex intracellular signaling pathways activated by the cytokines TNF‐α, IL‐1, the prostaglandin, PGE‐2, and epidermal growth factor (EGF) that regulate this gene is shown in Figure 37.3. A large number of potential binding sites for transcription factors have been identified along the promoter region of the collagen I gene, and additional regulatory sites have also been found in the first exon of the gene. Although the regulation of the collagen gene via these transcription factors is complex, and is not completely understood, several features of the regulatory sites in the promoter region have been characterized. Figure 37.3 Regulation of the collagen I gene by inflammatory mediators and by vitamin C. Sites marked with a gray/dark green circle with “Inh” inside the circle are pathways that turn OFF collagen gene transcription. Sites marked with a yellow circle with a red X inside the circle are pathways that vitamin C blocks, thereby preventing the inhibition of the collagen gene. See text for more detail. It is somewhat surprising that there are so many hormone‐like mediators that suppress collagen I gene expression, and so few that stimulate transcription. TNF‐α, IL‐1, EGF, Interferon, FGF, and PGE‐2 all inhibit the gene. Additionally, retinoids [83] and 1,25 dihydroxyvitamin D [84] have also been shown to inhibit collagen I transcription. The list of mediators that stimulate the gene is quite short, comprising primarily TGF β [78], vitamin C [62, 85], and IL‐4 [59]. Fortunately, because of its bi‐functional activity, vitamin C has the ability to block some of the signaling pathways used by cytokines and growth factors to suppress collagen I gene activity. As shown in Figure 37.3, vitamin C can block the TNF‐α [38] and IL‐1 [86] mediated inhibition of collagen I gene transcription by preventing the phosphorylation of IKB by IKK [39, 87]. Unless it is phosphorylated, IKB cannot dissociate from NF‐kB, and this prevents NF‐kB activation. Thus, the inhibitory effect of Nf‐kB on the collagen I gene promoter is prevented. Similarly, vitamin C can prevent the activation of the MAPK/ERK by cytokines and as a result the transcription factor C/EBP is not phosphorylated, and cannot suppress the collagen gene [88]. Although vitamin C can increase collagen gene transcription by about fourfold, and can also block the repression of the gene by various cytokines, unfortunately, it cannot block all cytokines and growth factors. For example, vitamin C cannot prevent the inhibition of collagen I gene by EGF [71, 72], even though it can inhibit the ERK pathway activated by other cytokines and growth factors. It is also ineffective in preventing the PGE‐2‐mediated repression of the gene [89]. Nevertheless, by being able to: (1) inactivate ROS, (2) stimulate collagen I gene transcription, and (3) block signaling pathways that cause gene suppression, vitamin C plays an important role in maintaining high levels of collagen in the skin. Since the inflammatory process in skin is extremely complex and involves the production and action of many cytokines and chemokines, as well as various paracrine and autocrine factors, how does one decide which anti‐inflammatory compound to use in a topical formulation to achieve the best efficacy in treating skin problems, including aging? Of all the inflammatory cytokines, chemokines and other hormone‐like mediators produced in the skin in response to environmental stressors like UVR and pollutants, there are three mediators that play significant roles in skin diseases and in skin aging. These three are TNF‐α, IL‐1, and PGE‐2. Designing therapeutics that contain natural “bi‐functional” antioxidants that can block all three of these inflammatory mediators would not only provide effective relief for many inflammatory skin problems, but would also likely provide benefits for reducing both photoaging and chronological skin aging. One of the most potent natural “bi‐functional” anti‐inflammatory antioxidants may be curcumin. In addition to its ability to neutralize free radicals, it can also interfere with cell signaling pathways involved in inflammation. Considerable research efforts have focused on understanding how curcumin blocks inflammatory pathways [112–115]. From these studies, the following picture has emerged: In addition to curcumin, a large number of other phenolic and polyphenolic compounds derived from plants have now been shown to have direct anti‐inflammatory effects on cells as well as antioxidant activity. These include quercetin [120], luteolin [121], resveratrol [122, 123], 4‐propyl guaiacol [105], ferulic acid [124], apigenin [125], genistein [126], and epigallocatechin gallate [127, 128]. While some of these antioxidants reduce inflammatory gene expression through interfering with NF‐κB‐driven genes, others interfere with MAPK signaling pathways that lead to the activation of the transcription factor, AP‐1. By interfering with the ability of AP‐1 to activate target genes, these compounds, such as quercetin and luteolin, prevent the activation of such inflammatory genes as IL‐6, COX‐2, IL‐18, and MCP‐1 (monocyte chemotactic protein‐1) [129–132]. With regard to skin anti‐aging benefits, not only can luteolin reduce the expression of inflammatory cytokines and chemokines, but it also inhibits hyaluronidases and thus preserves hyaluronic acid levels in the skin [133]. A related polyphenol, genistein, blocks intracellular signaling pathways by inhibiting the phosphorylation of the epidermal growth factor receptor and the activation of the receptor‐associated tyrosine kinase (EGF‐R) [126]. Figure 37.4 shows a few of the inflammatory mediators that several of these phenolic bi‐functional antioxidants can inhibit in fibroblasts, keratinocytes, and monocytes. Because many of the phenolics that show the strongest anti‐inflammatory activity are bright yellow in color, their use in topical products has been limited. However, a derivative of curcumin, tetrahydrocurcumin, has essentially the same bioactivity as curcumin, but is colorless. Panel D in Figure 37.4 shows some of the inhibitory effects of tetrahydrocurcumin on the production of and/or activity of several inflammatory mediators.
CHAPTER 37
Antioxidants and Anti‐inflammatories
Antioxidants
Introduction
Antioxidants, free radicals, and reactive oxygen species (ROS)
ROS effects on signaling pathways
ROS and glycation: effects on skin aging
Cell type
Action
Aging effect
Keratinocyte
Decreased proliferation
Reduced cell renewal
Keratinocyte
Increased MMPs
Matrix destruction
Keratinocyte
NFκB activation
Inflammation
Fibroblasts
Decreased proliferation
Increased senescence
Fibroblasts
MMP increase decreased collagen synthesis
Loss of matrix strength and elasticity
Fibroblasts
Increased apoptosis
Cell death, thinning skin
Fibroblasts
Increased ROS
Inflammation
Vascular endothelium
Increase adhesion molecules in endothelial cells
Increase in immune cells into skin: increased cytokines and chemokines. Inflammation 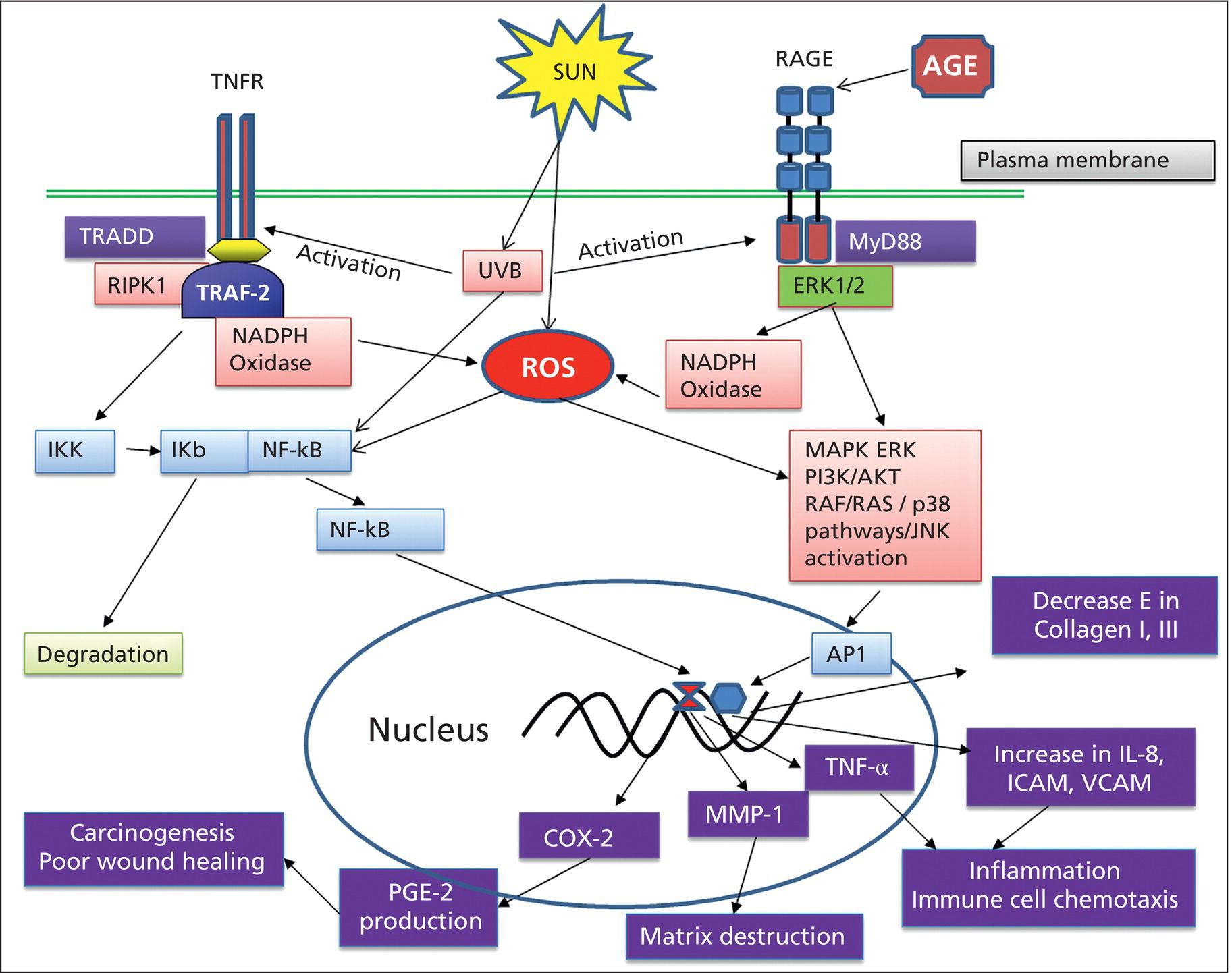
Antioxidants protect cells from free radicals, ROS, and glycation
Antioxidant
ORAC mole TE/100 g
Olive leaf extract
6,757,000
EGCG
2,285,714
Ferulic acid
1,941,700
Curcumin
1,500,000
Clove
1,078,700
Quercetin
1,090,000
Vanillin
1,046,052
4‐Ethyl guaiacol
1,029,630
Resveratrol
620,000
Cinnamon
267,536
Vitamin C
189,000
Vitamin E
135,000
Acai
102,700
Cocoa
80,7000
Pomegranate
10,500
Blueberry (raw)
7,700
Lycopene
5,800
Biology of the skin inflammatory process
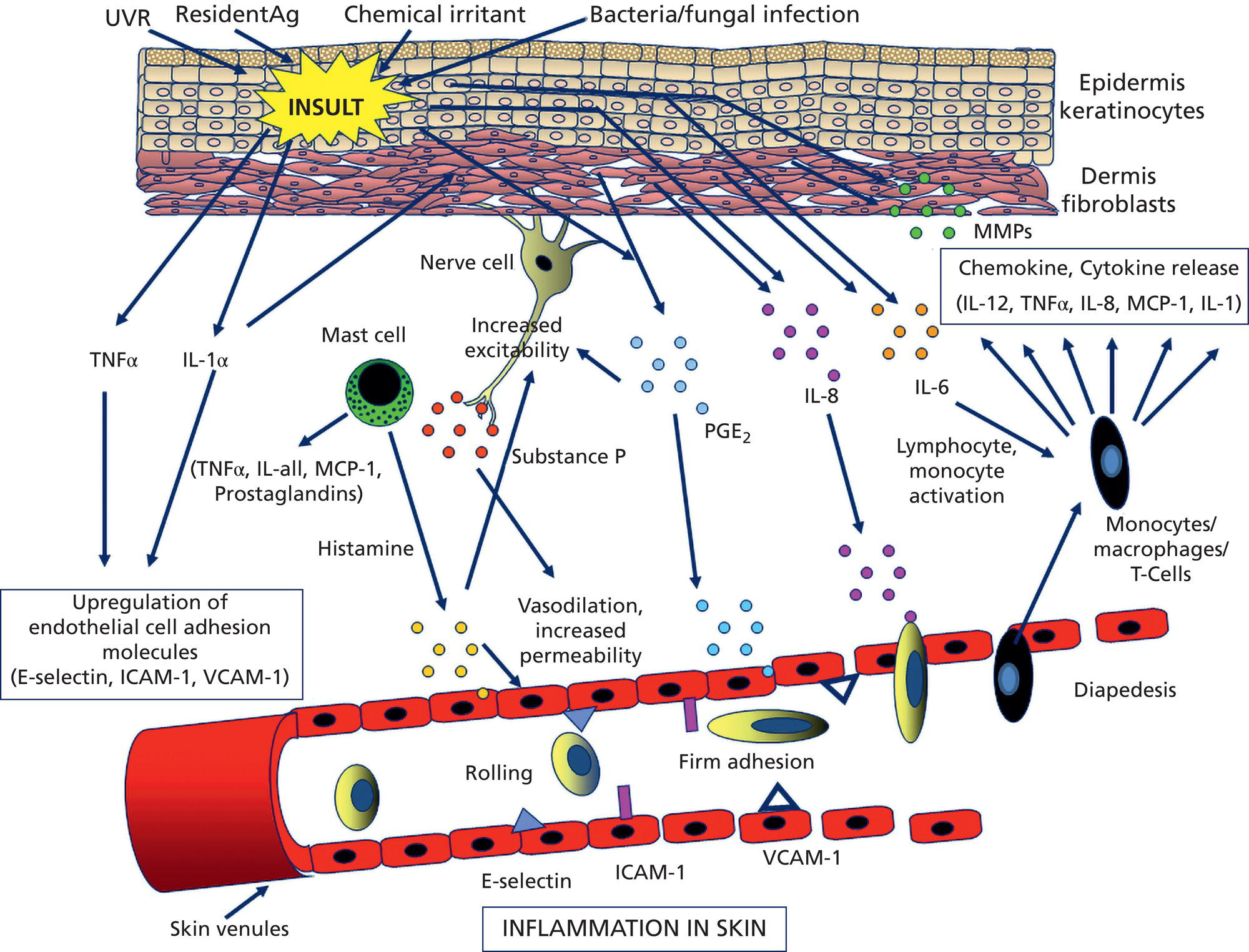
Anti‐inflammatory activities of bi‐functional antioxidants: effects on cell signaling pathways
Developing effective skin care therapeutics by targeting three key inflammatory mediators
Can oral antioxidants alter skin function?
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


